Autoimmune hemolytic anemia
| Autoimmune hemolytic anemia | |
|---|---|
| Other names: Autoimmune haemolytic anaemia | |
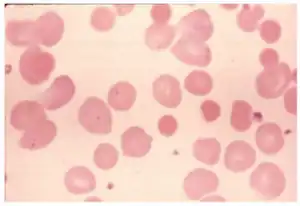 | |
| Idiopathic autoimmune hemolytic anemia because of lecithin overdose-peripheral blood smear shows spherocytes | |
| Specialty | Hematology |
Autoimmune hemolytic anemia (AIHA) occurs when antibodies directed against the person's own red blood cells (RBCs) cause them to burst (lyse), leading to an insufficient number of oxygen-carrying red blood cells in the circulation. The lifetime of the RBCs is reduced from the normal 100–120 days to just a few days in serious cases.[1][2] The intracellular components of the RBCs are released into the circulating blood and into tissues, leading to some of the characteristic symptoms of this condition. The antibodies are usually directed against high-incidence antigens, therefore they also commonly act on allogenic RBCs (RBCs originating from outside the person themselves, e.g. in the case of a blood transfusion).[3] AIHA is a relatively rare condition, with an incidence of 5-10 cases per 1 million persons per year in the warm-antibody type and 0.45 to 1.9 cases per 1 million persons per year in the cold antibody type.[4] Autoimmune hemolysis might be a precursor of later onset systemic lupus erythematosus.[5]
The terminology used in this disease is somewhat ambiguous. Although MeSH uses the term "autoimmune hemolytic anemia",[6] some sources prefer the term "immunohemolytic anemia" so drug reactions can be included in this category.[7][8] The National Cancer Institute considers "immunohemolytic anemia", "autoimmune hemolytic anemia", and "immune complex hemolytic anemia" to all be synonyms.[9]
Signs and symptoms
Symptoms of AIHA may be due to the underlying anemia; including shortness of breath or dyspnea, fatigue, headache, muscle weakness and pallor.[10] In cold agglutinin disease (cold antibody type), agglutination and impaired passage of red blood cells through capillaries in the extremities causes acrocyanosis and Raynaud phenomenon with a rare complication of gangrene[4]
Spherocytes are found in immunologically-mediated hemolytic anemias.[11] Signs of hemolysis that are present in AIHA include low hemoglobin (blood count), alterations in levels of cell markers of hemolysis; including elevated lactate dehydrogenase (LDH), decreased haptoglobin and elevated unconjugated bilirubin.[4] Reticulocytosis, or an increase in circulating immature red blood cells, may be seen.[4]
Causes
The causes of AIHA are poorly understood. The disease may be primary, or secondary to another underlying illness. The primary illness is idiopathic (the two terms used synonymously). Idiopathic AIHA accounts for approximately 50% of cases.[12][13]
Secondary AIHA can result from many other illnesses. Warm and cold type AIHA each have their own more common secondary causes. The most common causes of secondary warm-type AIHA include lymphoproliferative disorders (e.g., chronic lymphocytic leukemia, lymphoma) and other autoimmune disorders (e.g., systemic lupus erythematosus, rheumatoid arthritis, scleroderma, Crohn's disease, ulcerative colitis). Less common causes of warm-type AIHA include neoplasms other than lymphoid, and infection. Secondary warm type AIHA has been observed in cases of Covid-19.[4] Secondary cold type AIHA is also caused primarily by lymphoproliferative disorders but is also commonly caused by infection, especially by mycoplasma, viral pneumonia, infectious mononucleosis, and other respiratory infections. Less commonly, it can be caused by concomitant autoimmune disorders.[14]
Drug-induced AIHA, though rare, can be caused by a number of drugs, including α-methyldopa and penicillin. This is a type II immune response in which the drug binds to macromolecules on the surface of the RBCs and acts as an antigen. Antibodies are produced against the RBCs, which leads to complement activation. Complement fragments, such as C3a, C4a and C5a, activate granular leukocytes (e.g., neutrophils), while other components of the system (C6, C7, C8, C9) either can form the membrane attack complex (MAC) or can bind the antibody, aiding phagocytosis by macrophages (C3b). This is one type of "penicillin allergy".
In about half of cases, the cause of autoimmune hemolytic anemia cannot be determined (idiopathic or primary). This condition can also be caused by or occur with another disorder (secondary) or rarely, occur following the use of certain drugs (such as penicillin) or after a person has a blood and marrow stem cell transplant.[15]
Secondary causes of autoimmune hemolytic anemia include:[15]
Pathophysiology
AIHA can be caused by a number of different classes of antibody, with IgG and IgM antibodies being the main causative classes. Depending on which is involved, the pathology will differ. IgG is not very effective at activating complement and effectively binds the Fc receptor (FcR) of phagocytic cells,[16] AIHA involving IgG is generally characterized by phagocytosis of RBCs. IgM is a potent activator of the classical complement pathway, thus, AIHA involving IgM is characterized by complement-mediated lysis of RBCs. IgM also leads to phagocytosis of RBCs however, because phagocytic cells have receptors for the bound complement (rather than FcRs as in IgG AIHA). In general, IgG AIHA takes place in the spleen, whereas IgM AIHA takes place in Kupffer cells – phagocytic cells of the liver. Phagocytic AIHA is termed extravascular, whereas complement-mediated lysis of RBCs is termed intravascular AIHA. In order for intravascular AIHA to be recognizable, it requires overwhelming complement activation, therefore most AIHA is extravascular – be it IgG- or IgM-mediated.[3]
AIHA cannot be attributed to any single autoantibody. To determine the autoantibody or autoantibodies present in a patient, the Coombs test, also known as the antiglobulin test, is performed. There are two types of Coombs tests, direct and indirect; more commonly, the direct antiglobulin test (DAT) is used. Classification of the antibodies is based on their activity at different temperatures and their etiology. Antibodies with high activity at physiological temperature (approximately 37 °C) are termed warm autoantibodies. Cold autoantibodies act best at temperatures of 0–4 °C. Patients with cold-type AIHA, therefore, have higher disease activity when body temperature falls into a hypothermic state. Usually, the antibody becomes active when it reaches the limbs, at which point it opsonizes RBCs. When these RBCs return to central regions, they are damaged by complement. Patients may present with one or both types of autoantibodies; if both are present, the disease is termed "mixed-type" AIHA.
When DAT is performed, the typical presentations of AIHA are as follows. Warm-type AIHA shows a positive reaction with antisera to IgG antibodies with or without complement activation. Cases may also arise with complement alone or with IgA, IgM or a combination of these three antibody classes and complement. Cold-type AIHA usually reacts with antisera to complement and occasionally to the above antibodies. This is the case in both cold agglutinin disease and cold paroxysmal hematuria. In general, mixed warm and cold AIHA shows a positive reaction to IgG and complement, sometimes IgG alone, and sometimes complement alone. Mixed-type can, like the others, present unusually with positive reactions to other antisera.[14]
Diagnosis
Diagnosis is made by first ruling out other causes of hemolytic anemia, such as G6PD, thalassemia, sickle-cell disease. Clinical history is also important to elucidate any underlying illness or medications that may have led to the disease.
Laboratory investigations are carried out to determine the etiology of the disease. Following confirmation of hemolysis (seen with laboratory markers of low hemoglobin, elevated LDH, decreased haptoglobin, and elevated unconjugated bilirubin), a direct antiglobulin test (DAT)(also known as a Coomb's test) is done to show auto-immune pathogenesis with antibodies, compliment or both on the erythrocyte surface.[4] This is followed by a monospecific DAT that identifies the specific antibody and compliment types on the erythrocyte surface.[4] In cold agglutinin disease, the monospecific DAT is by definition positive for the compliment molecule C3d but IgM may be negative as the molecule may detach at the time of testing.[4] The diagnosis of cold agglutinin disease is confirmed with an elevated cold agglutinin titer.[4]
A bone marrow biopsy is used in AIHA to identify a possible underlying lymphoproliferative disorder.[4]
Classification
AIHA can be classified as warm autoimmune hemolytic anemia or cold autoimmune hemolytic anemia, which includes cold agglutinin disease and paroxysmal cold hemoglobinuria. These classifications are based on the characteristics of the autoantibodies involved in the pathogenesis of the disease. Each has a different underlying cause, management, and prognosis, making classification important when treating a patient with AIHA.[17]
- Autoimmune hemolytic anemia
-
- Primary
- Secondary (lymphoproliferative disorders, autoimmune disorders)[18]: 259
- Primary cold agglutinin disease
- Secondary cold agglutinin syndrome
- Associated with malignant disease
- Acute, transient,[18] infection-associated (acute cold antibody mediated AIHA complicating Mycoplasma pneumoniae or viral infections[19])
- Chronic (lymphoproliferative disorders)[18]: 259
- Idiopathic
- Secondary
- Idiopathic
- Secondary (lymphoproliferative disorders, autoimmune disorders)[18]
- Drug-induced immune hemolytic anemia[18]: 259
- Autoimmune type
- Drug absorption type
- Neoantigen type[20]
-
Evidence for hemolysis
The following findings may be present:[21][22]
- Increased red cell breakdown
- Elevated serum bilirubin (unconjugated)
- Excess urinary urobilinogen
- Reduced plasma haptoglobin
- Raised serum lactic dehydrogenase (LDH)
- Hemosiderinuria
- Methemalbuminemia
- Spherocytosis
- Increased red cell production:
- Reticulocytosis
- Erythroid hyperplasia of the bone marrow
- Specific investigations
- Positive direct Coombs test
Treatment
Steroids are the first line treatment in warm AIHA; with oral prednisone achieving an 80% initial response rate, with a 30-40% sustained remission rate at 1 year.[4] Steroids may be decreased at 3 weeks and tapered at 3-6 months depending on the response.[4] Rituximab may be added to initial management to increase the response rate, or it may be used in cases of severe disease such as IgA mediated warm AIHA, mixed AIHA, Evans syndrome or in cases of high hemolysis levels).[4] If a response cannot be achieved with steroids or rituximab, splenectomy can be done.[4] Other third line options, that are less studied, include azathioprine, cyclophosphamide, cyclosporine, mycophenolate mofetil and bortezomib.[4] The treatments for secondary warm AIHA are generally the same as primary warm AIHA, but with the addition of treating the underlying disease if possible.[4]
Steroids are not indicated in the treatment of cold agglutinin disease due to low response rates.[4] Cases of cold agglutinin disease with mild anemia with limited and compensated hemolysis can be monitored with adjunct supportive care (such as avoidance of cold exposure or thermal protection to prevent against hemolysis).[4] Rituximab is used to treat pathogenic B-cell clones in cold agglutinin disease with response rates of 45-60%.[4] Relapses are common upon discontinuation of rituximab, but the medication can be restarted to achieve subsequent remission.[4] Rituximab can be combined with bendamustine to achieve a 71% overall and 40% complete response rate with an increased response seen with prolonged therapy (with a time to best response at a median of 30 months) due to the drugs' effect on long lived plasma cells.[4] Splenectomy is less efficacious in cold agglutinin disease.[23]
Special considerations are required when treating people with AIHA using blood transfusion. In cold agglutinin disease; the patient and the extremity should be kept warm during transfusion to prevent agglutination and hemolysis of the donor and patient red blood cells.[4] In warm AIHA; cross-matching of blood will show incompatibility so it is recommended to perform a bedside in vivo compatibility test prior to infusion.[4] Erythropoietin (EPO) has been shown to increase hemoglobin levels in cold and warm AIHA.[4]
History
"Blood-induced icterus" produced by the release of massive amounts of a coloring material from blood cells followed by the formation of bile was recognized and described by Vanlair and Voltaire Masius' in 1871. About 20 years later, Hayem distinguished between congenital hemolytic anemia and an acquired type of infectious icterus associated with chronic splenomegaly. In 1904, Donath and Landsteiner suggested a serum factor was responsible for hemolysis in paroxysmal cold hemoglobinuria. French investigators led by Chauffard stressed the importance of red-cell autoagglutination in patients with acquired hemolytic anemia. In 1930, Lederer and Brill described cases of acute hemolysis with rapid onset of anemia and rapid recovery after transfusion therapy. These hemolytic episodes were thought to be due to infectious agents. A clear distinction between congenital and acquired hemolytic anemia was not drawn, however, until Dameshek and Schwartz in 1938, and, in 1940, they demonstrated the presence of abnormal hemolysins in the sera of patients with acquired hemolytic anemia and postulated an immune mechanism.
During the past three decades, studies defining red-cell blood groups and serum antibodies have produced diagnostic methods that have laid the basis for immunologic concepts relevant to many of the acquired hemolytic states. Of these developments, the antiglobulin test described by Coombs, Mourant, and Race in 1945 has proved to be one of the more important, useful tools now available for the detection of immune hemolytic states. This technique demonstrated that a rabbit antibody against human globulin would induce agglutination of human red cells "coated with an incomplete variety of rhesus antibodies". C. Moreschlit had used the same method in 1908 in a goat antirabbit-red-cell system. The test was premature and was forgotten. In 1946, Boorman, Dodd, and Loutit applied the direct antiglobulin test to a variety of hemolytic anemias, and laid the foundation for the clear distinction of autoimmune from congenital hemolytic anemia.
A hemolytic state exists whenever the red cell survival time is shortened from the normal average of 120 days. Hemolytic anemia is the hemolytic state in which anemia is present, and bone marrow function is inferentially unable to compensate for the shortened lifespan of the red cell. Immune hemolytic states are those, both anemic and nonanemic, which involve immune mechanisms consisting of antigen-antibody reactions. These reactions may result from unrelated antigen-antibody complexes that fix to an innocent-bystander erythrocyte, or from related antigen-antibody combinations in which the host red cell or some part of its structure is or has become antigenic. The latter type of antigen-antibody reaction may be termed "autoimmune", and hemolytic anemias so produced are autoimmune hemolytic anemias.[2]
In children
In general, AIHA in children has a good prognosis and is self-limiting. However, if it presents within the first two years of life or in the teenage years, the disease often follows a more chronic course, requiring long-term immunosuppression, with serious developmental consequences. The aim of therapy may sometimes be to lower the use of steroids in the control of the disease. In this case, splenectomy may be considered, as well as other immunosuppressive drugs. Infection is a serious concern in patients on long-term immunosuppressant therapy, especially in very young children (less than two years).[24]
See also
References
- ↑ Shoenfield, Y; et al. (2008). Diagnostic Criteria in Autoimmune Disease. Humana Press.
- 1 2 Sawitsky A, Ozaeta PB (June 1970). "Disease-associated autoimmune hemolytic anemia". Bull N Y Acad Med. 46 (6): 411–26. PMC 1749710. PMID 5267234.
- 1 2 Gehrs BC, Friedberg RC (April 2002). "Autoimmune hemolytic anemia". Am. J. Hematol. 69 (4): 258–71. doi:10.1002/ajh.10062. PMID 11921020. S2CID 22547733.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Berentsen, Sigbjørn; Barcellini, Wilma (7 October 2021). "Autoimmune Hemolytic Anemias". New England Journal of Medicine. 385 (15): 1407–1419. doi:10.1056/NEJMra2033982.
- ↑ Kokori, SI; Ioannidis, JP; Voulgarelis, M; Tzioufas, AG; Moutsopoulos, HM (2000-02-15). "Autoimmune hemolytic anemia in patients with systemic lupus erythematosus". The American Journal of Medicine. 108 (3): 198–204. doi:10.1016/S0002-9343(99)00413-1. ISSN 0002-9343. PMID 10723973.
- ↑ Autoimmune+hemolytic+anemia at the US National Library of Medicine Medical Subject Headings (MeSH)
- ↑ Wright MS (1999). "Drug-induced hemolytic anemias: increasing complications to therapeutic interventions". Clin Lab Sci. 12 (2): 115–8. PMID 10387489.
- ↑ Cotran, Ramzi S.; Kumar, Vinay; Fausto, Nelson; Nelso Fausto; Robbins, Stanley L.; Abbas, Abul K. (2005). Robbins and Cotran pathologic basis of disease. St. Louis, Mo: Elsevier Saunders. p. 636. ISBN 978-0-7216-0187-8.
- ↑ "Definition of immunohemolytic anemia". NCI Dictionary of Cancer Terms. Archived from the original on 15 January 2009. Retrieved 2009-02-07.
- ↑ "Autoimmune hemolytic anemia | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Archived from the original on 2022-01-31. Retrieved 2022-01-29.
- ↑ Thoma J, Kutter D, Casel S, et al. (2005). "HbSC hemoglobinopathy suspected by chest x-ray and red blood cell morphology". Acta Clin Belg. 60 (6): 377–82. doi:10.1179/acb.2005.057. PMID 16502600. S2CID 43340793.
- ↑ Gupta S, Szerszen A, Nakhl F, et al. (2011). "Severe refractory autoimmune hemolytic anemia with both warm and cold autoantibodies that responded completely to a single cycle of rituximab: a case report". J Med Case Rep. 5: 156. doi:10.1186/1752-1947-5-156. PMC 3096571. PMID 21504611.
- ↑ Bradencarter (28 December 2017). "About Hemolytic Anemia". hemolyticanemia.org. Archived from the original on 2 March 2018. Retrieved 28 December 2017.
- 1 2 Sokol RJ, Hewitt S, Stamps BK (June 1981). "Autoimmune haemolysis: an 18-year study of 865 cases referred to a regional transfusion centre". Br Med J (Clin Res Ed). 282 (6281): 2023–7. doi:10.1136/bmj.282.6281.2023. PMC 1505955. PMID 6788179.
- 1 2 3 "Autoimmune hemolytic anemia". Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. 2014-02-24. Archived from the original on 2022-01-31. Retrieved 2019-05-28.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ↑ Abramson N, Gelfand EW, Jandl JH, Rosen FS (December 1970). "The interaction between human monocytes and red cells. Specificity for IgG subclasses and IgG fragments". J. Exp. Med. 132 (6): 1207–15. doi:10.1084/jem.132.6.1207. PMC 2180500. PMID 5511570.
- ↑ Zanella, A.; Barcellini, W. (2014-09-30). "Treatment of autoimmune hemolytic anemias". Haematologica. Ferrata Storti Foundation (Haematologica). 99 (10): 1547–1554. doi:10.3324/haematol.2014.114561. ISSN 0390-6078. PMC 4181250. PMID 25271314.
- 1 2 3 4 5 6 7 8 Gehrs, B. C.; Friedberg, R. C. (2002). "Concise review: Autoimmune Hemolytic Anemia". American Journal of Hematology. Wiley. 69 (4): 258–71. doi:10.1002/ajh.10062. PMID 11921020. S2CID 22547733.
- ↑ Berentsen, Sigbjørn; Beiske, Klaus; Tjønnfjord, Geir E. (2007-07-21). "Primary chronic cold agglutinin disease: An update on pathogenesis, clinical features and therapy". Hematology (Amsterdam, Netherlands). Informa UK Limited. 12 (5): 361–370. doi:10.1080/10245330701445392. ISSN 1607-8454. PMC 2409172. PMID 17891600.
- ↑ Berentsen, Sigbjørn; Sundic, Tatjana (2015-01-29). "Red Blood Cell Destruction in Autoimmune Hemolytic Anemia: Role of Complement and Potential New Targets for Therapy". BioMed Research International. Hindawi Limited. 2015. 363278-1–363278-11. doi:10.1155/2015/363278. ISSN 2314-6133. PMC 4326213. PMID 25705656.
- ↑ Kumar P, Clark M (2005). Clinical Medicine (6th ed.). Elsevier Saunders. p. 437.
- ↑ "Autoimmune Hemolytic Anemia". The Lecturio Medical Concept Library. Archived from the original on 9 July 2021. Retrieved 3 July 2021.
- ↑ Go R. S., Winters J. L., Kay N. E. (2017). "How I treat autoimmune hemolytic anemia". Blood. 129 (22): 2971–2979. doi:10.1182/blood-2016-11-693689. PMID 28360039.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ↑ Zecca M, Nobili B, Ramenghi U, et al. (May 2003). "Rituximab for the treatment of refractory autoimmune hemolytic anemia in children". Blood. 101 (10): 3857–61. doi:10.1182/blood-2002-11-3547. PMID 12531800.
External links
| Classification | |
|---|---|
| External resources |
|