Robinow syndrome
| Robinow syndrome | |
|---|---|
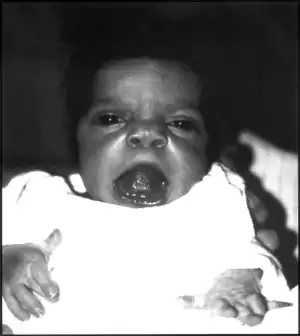 | |
| An infant exhibiting the facial features of Robinow syndrome. | |
| Specialty | Medical genetics |
Robinow syndrome is an extremely rare genetic disorder characterized by short-limbed dwarfism, abnormalities in the head, face, and external genitalia, as well as vertebral segmentation. The disorder was first described in 1969 by human geneticist Meinhard Robinow,[1] along with physicians Frederic N. Silverman and Hugo D. Smith, in the American Journal of Diseases of Children. By 2002, over 100 cases had been documented and introduced into medical literature.[1]
Two forms of the disorder exist, dominant and recessive, of which the former is more common. Patients with the dominant version often suffer moderately from the aforementioned symptoms. Recessive cases, on the other hand, are usually more physically marked, and individuals may exhibit more skeletal abnormalities.[2] The recessive form is particularly frequent in Turkey.[3] However, this can likely be explained by a common ancestor, as these patients' families can be traced to a single town in Eastern Turkey.[4] Clusters of the autosomal recessive form have also been documented in Oman and Czechoslovakia.[1]
The syndrome is also known as Robinow-Silverman-Smith syndrome, Robinow dwarfism, fetal face, fetal face syndrome,[5] fetal facies syndrome, acral dysostosis with facial and genital abnormalities, or mesomelic dwarfism-small genitalia syndrome.[6] The recessive form was previously known as Covesdem syndrome.
Signs and symptoms

Robinow noted the resemblance of affected patients' faces to that of a fetus, using the term "fetal facies" to describe the appearance of a small face and widely spaced eyes.[1] Clinical features also may include a short, upturned nose, a prominent forehead, and a flat nasal bridge. The upper lip may be "tented",[1] exposing dental crowding, "tongue tie", or gum hypertrophy.
Though the eyes do not protrude, abnormalities in the lower eyelid may give that impression. Surgery may be necessary if the eyes cannot close fully. In addition, the ears may be set low on the head or have a deformed pinna.[1]
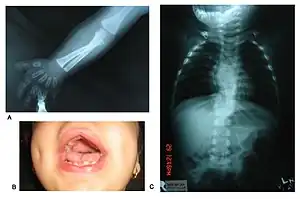
Patients suffer from dwarfism, short lower arms, small feet, and small hands. Fingers and toes may also be abnormally short and laterally or medially bent. The thumb may be displaced and some patients, notably in Turkey, experience ectrodactyly.[1] All patients often suffer from vertebral segmentation abnormalities. Those with the dominant variant have, at most, a single butterfly vertebra.[2] Those with the recessive form, however, may suffer from hemivertebrae, vertebral fusion, and rib anomalies. Some cases resemble Jarcho-Levin syndrome or spondylocostal dysostosis.[1]
Genital defects characteristically seen in males include a micropenis with a normally developed scrotum and testes. Sometimes, testicles may be undescended, or the patient may suffer from hypospadias.[2] Female genital defects may include a reduced size clitoris and underdeveloped labia minora. Infrequently, the labia majora may also be underdeveloped.[2] Some research has shown that females may experience vaginal atresia or haematocolpos.[3]
The autosomal recessive form of the disorder tends to be much more severe. Examples of differences are summarized in the following table:[7]
| Characteristic | Autosomal recessive | Autosomal dominant |
|---|---|---|
| Stature | Shorter stature -2 SD or less | Short or normal |
| Arms | Very short | Slightly short |
| Elbow | Radial head dislocation | No radial head dislocation |
| Upper lip | Tented upper lip | Normal upper lip |
| Mortality rate | 10% mortality | No excess mortality |
Associated conditions
Medical conditions include frequent ear infection, hearing loss, hypotonia, developmental problems, respiratory problems, eating difficulties, light sensitivity, and esophageal reflux.[2]
Data on fertility and the development of secondary sex characteristics is relatively sparse. It has been reported that both male and female patients have had children. Males who have reproduced have all had the autosomal dominant form of the disorder; the fertility of those with the recessive variant is unknown.[1]
Researchers have also reported abnormalities in the renal tract of affected patients. Hydronephrosis is a relatively common condition, and researchers have theorized that this may lead to urinary tract infections.[8] In addition, a number of patients have suffered from cystic dysplasia of the kidney.[1]
A number of other conditions are often associated with Robinow syndrome. About 15% of reported patients suffer from congenital heart defects. Though there is no clear pattern, the most common conditions include pulmonary stenosis and atresia.[9] In addition, though intelligence is generally normal, around 15% of patients show developmental delays.[1]
Genetics
Genetic studies have linked the autosomal recessive form of the disorder to the ROR2 gene on position 9 of the long arm of chromosome 9.[1] The gene is responsible for aspects of bone and cartilage growth. This same gene is involved in causing autosomal dominant brachydactyly B.[1]
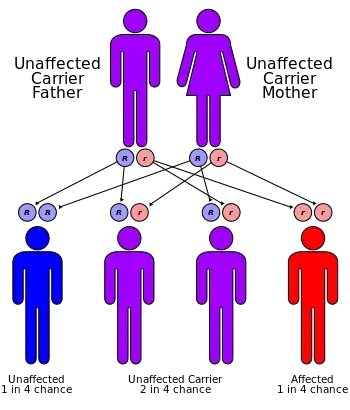
The autosomal dominant form has been linked to three genes - WNT5A, Segment polarity protein dishevelled homolog DVL-1 (DVL1) and Segment polarity protein dishevelled homolog DVL-3 (DVL3). This form is often caused by new mutations and is generally less severe than the recessive form. Two further genes have been linked to this disorder - Frizzled-2 (FZD2) and Nucleoredoxin (NXN gene).[10] All of these genes belong to the same metabolic pathway - the WNT system. This system is involved in secretion for various compounds both in the fetus and in the adult.
A fetal ultrasound can offer prenatal diagnosis 19 weeks into pregnancy. However, the characteristics of a fetus suffering from the milder dominant form may not always be easy to differentiate from a more serious recessive case. Genetic counseling is an option given the availability of a family history.[1]
Diagnosis
Robinow syndrome is suspected by clinical findings and family history and confirmed by typical ROR-2 biallelic pathogenic variants identified by molecular genetic testing.[11]
Treatment
Treatment of the various manifestations will usually be addressed by a multidisciplinary team.[12]
History
The disorder was first described in 1969 by the German-American Human Geneticist Meinhard Robinow (1909–1997),[1] along with physicians Frederic N. Silverman and Hugo D. Smith, in the American Journal of Diseases of Children. By 2002, over 100 cases had been documented and introduced into medical literature.[1]
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 Patton, M A; Afzal, A. R (2002). "Robinow syndrome". Journal of Medical Genetics. 39 (5): 305–10. doi:10.1136/jmg.39.5.305. PMC 1735132. PMID 12011143.
- 1 2 3 4 5 Robinow Syndrome Foundation. General Information. Accessed 19 May 2006.
- 1 2 Balci, Sevim; Beksaç, Sinan; Haliloglu, Mithat; Ercis, Murat; Eryilmaz, Muzaffer (1998). "Robinow syndrome, vaginal atresia, hematocolpos, and extra middle finger". American Journal of Medical Genetics. 79 (1): 27–9. doi:10.1002/(SICI)1096-8628(19980827)79:1<27::AID-AJMG7>3.0.CO;2-F. PMID 9738864.
- ↑ Brunner, Han G; Van Bokhoven, Hans; Celli, Jacopo; Kayserili, Hülya; Van Beusekom, Ellen; Balci, Sevim; Brussel, Wim; Skovby, Flemming; Kerr, Bronwyn; Percin, E. Ferda; Akarsu, Nurten (2000). "Mutation of the gene encoding the ROR2 tyrosine kinase causes autosomal recessive Robinow syndrome". Nature Genetics. 25 (4): 423–6. doi:10.1038/78113. PMID 10932187. S2CID 36402844.
- ↑ National Organization for Rare Disorders, Inc. Robinow Syndrome. Last modified 15 May 2006. Accessed 19 May 2006.
- ↑ Jablonski's Syndromes Database. Multiple Congenital Anomaly/Mental Retardation (MCA/MR) Syndromes. Accessed 20 May 2006.
- ↑ Robinow, M (1993). "The Robinow (fetal face) syndrome". Clinical Dysmorphology. 2 (3): 189–98. doi:10.1097/00019605-199307000-00001. PMID 8287180. S2CID 38507817.
- ↑ Shprintzen, Robert J; Goldberg, R. B; Saenger, P; Sidoti, E. J (1982). "Male-to-Male Transmission of Robinow's Syndrome". American Journal of Diseases of Children. 136 (7): 594–7. doi:10.1001/archpedi.1982.03970430026007. PMID 7091086.
- ↑ Webber, Steven A; Wargowski, David S; Chitayat, David; Sandor, George G. S (1990). "Congenital heart disease and Robinow syndrome: Coincidence or an additional component of the syndrome?". American Journal of Medical Genetics. 37 (4): 519–21. doi:10.1002/ajmg.1320370418. PMID 2260599.
- ↑ White, Janson J; Mazzeu, Juliana F; Coban-Akdemir, Zeynep; Bayram, Yavuz; Bahrambeigi, Vahid; Hoischen, Alexander; Van Bon, Bregje W.M; Gezdirici, Alper; Gulec, Elif Yilmaz; Ramond, Francis; Touraine, Renaud; Thevenon, Julien; Shinawi, Marwan; Beaver, Erin; Heeley, Jennifer; Hoover-Fong, Julie; Durmaz, Ceren D; Karabulut, Halil Gurhan; Marzioglu-Ozdemir, Ebru; Cayir, Atilla; Duz, Mehmet B; Seven, Mehmet; Price, Susan; Ferreira, Barbara Merfort; Vianna-Morgante, Angela M; Ellard, Sian; Parrish, Andrew; Stals, Karen; Flores-Daboub, Josue; et al. (2018). "WNT Signaling Perturbations Underlie the Genetic Heterogeneity of Robinow Syndrome". The American Journal of Human Genetics. 102 (1): 27–43. doi:10.1016/j.ajhg.2017.10.002. PMC 5777383. PMID 29276006.
- ↑ Afzal AR, Jeffery S (July 2003). "One gene, two phenotypes: ROR2 mutations in autosomal recessive Robinow syndrome and autosomal dominant brachydactyly type B". Hum. Mutat. 22 (1): 1–11. doi:10.1002/humu.10233. PMID 12815588. S2CID 21096559.
- ↑ Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, Roifman M, Brunner H, Lohr J, Mazzeu J, Chitayat D (October 2019). Autosomal Dominant Robinow Syndrome in GeneReviews. PMID 25577943.
Further reading
- White, Janson; Mazzeu, Juliana F; Hoischen, Alexander; Jhangiani, Shalini N; Gambin, Tomasz; Alcino, Michele Calijorne; Penney, Samantha; Saraiva, Jorge M; Hove, Hanne; Skovby, Flemming; Kayserili, Hülya; Estrella, Elicia; Vulto-Van Silfhout, Anneke T; Steehouwer, Marloes; Muzny, Donna M; Sutton, V. Reid; Gibbs, Richard A; Lupski, James R; Brunner, Han G; Van Bon, Bregje W.M; Carvalho, Claudia M.B (2015). "DVL1 Frameshift Mutations Clustering in the Penultimate Exon Cause Autosomal-Dominant Robinow Syndrome". The American Journal of Human Genetics. 96 (4): 612–22. doi:10.1016/j.ajhg.2015.02.015. PMC 4385180. PMID 25817016.
External links
| Wikimedia Commons has media related to Robinow syndrome. |
- Disease ID 5704 at NIH's Office of Rare Diseases