MAPK/ERK pathway

The MAPK/ERK pathway (also known as the Ras-Raf-MEK-ERK pathway) is a chain of proteins in the cell that communicates a signal from a receptor on the surface of the cell to the DNA in the nucleus of the cell.
The signal starts when a signaling molecule binds to the receptor on the cell surface and ends when the DNA in the nucleus expresses a protein and produces some change in the cell, such as cell division. The pathway includes many proteins, such as mitogen-activated protein kinases (MAPKs), originally called extracellular signal-regulated kinases (ERKs), which communicate by adding phosphate groups to a neighboring protein (phosphorylating it), thereby acting as an "on" or "off" switch.
When one of the proteins in the pathway is mutated, it can become stuck in the "on" or "off" position, a necessary step in the development of many cancers. In fact, components of the MAPK/ERK pathway were first discovered in cancer cells, and drugs that reverse the "on" or "off" switch are being investigated as cancer treatments.[1]
Background
The signal that starts the MAPK/ERK pathway is the binding of extracellular mitogen to a cell surface receptor. This allows a Ras protein (a Small GTPase) to swap a GDP molecule for a GTP molecule, flipping the "on/off switch" of the pathway. The Ras protein can then activate MAP3K (e.g., Raf), which activates MAP2K, which activates MAPK. Finally, MAPK can activate a transcription factor, such as Myc. This process is described in more detail below.
Ras activation
Receptor-linked tyrosine kinases, such as the epidermal growth factor receptor (EGFR), are activated by extracellular ligands, such as the epidermal growth factor (EGF). Binding of EGF to the EGFR activates the tyrosine kinase activity of the cytoplasmic domain of the receptor. The EGFR becomes phosphorylated on tyrosine residues. Docking proteins such as GRB2 contain an SH2 domain that binds to the phosphotyrosine residues of the activated receptor.[2] GRB2 binds to the guanine nucleotide exchange factor SOS by way of the two SH3 domains of GRB2. When the GRB2-SOS complex docks to phosphorylated EGFR, SOS becomes activated.[3] Activated SOS then promotes the removal of GDP from a member of the Ras subfamily (most notably H-Ras or K-Ras). The Ras protein can then bind GTP and become active.
Apart from EGFR, other cell surface receptors that can activate this pathway via GRB2 include Trk A/B, Fibroblast growth factor receptor (FGFR) and PDGFR.
Kinase cascade
Activated Ras then activates the protein kinase activity of a RAF kinase.[4] The RAF kinase phosphorylates and activates a MAPK/ERK Kinase (MEK1 or MEK2). The MEK phosphorylates and activates a mitogen-activated protein kinase (MAPK).
RAF and MAPK/ERK are both serine/threonine-specific protein kinases. MEK is a serine/tyrosine/threonine kinase.
In a technical sense, RAF, MEK, and MAPK are all mitogen-activated kinases, as is MNK (see below). MAPKs were originally called "extracellular signal-regulated kinases" (ERKs) and "microtubule associated protein kinases" (MAPKs). One of the first proteins known to be phosphorylated by ERK was a microtubule-associated protein (MAP). As discussed below, many additional targets for phosphorylation by MAPK were later found, and the protein was renamed "mitogen-activated protein kinase" (MAPK). The series of kinases from RAF to MEK to MAPK is an example of a protein kinase cascade. Such series of kinases provide opportunities for feedback regulation and signal amplification.
Regulation of translation and transcription
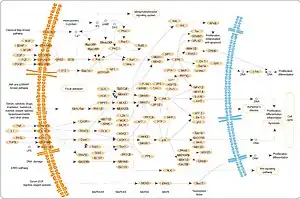
Three of the many proteins that are phosphorylated by MAPK are shown in the figure to the right. One effect of MAPK activation is to alter the translation of mRNA to proteins. MAPK phosphorylates the 40S ribosomal protein S6 kinase (RSK). This activates RSK, which, in turn, phosphorylates ribosomal protein S6.[5] Mitogen-activated protein kinases that phosphorylate ribosomal protein S6 were the first to be isolated.[4]
MAPK regulates the activities of several transcription factors. MAPK can phosphorylate C-myc. MAPK phosphorylates and activates MNK, which, in turn, phosphorylates CREB. MAPK also regulates the transcription of the C-Fos gene. By altering the levels and activities of transcription factors, MAPK leads to altered transcription of genes that are important for the cell cycle.
The 22q11, 1q42, and 19p13 genes, by affecting the ERK pathway, are associated with schizophrenia, schizoaffective disorder, bipolar disorder, and migraines.
Regulation of cell cycle entry and proliferation
Role of mitogen signaling in cell cycle progression The ERK pathway plays an important role of integrating external signals from the presence of mitogens such as epidermal growth factor (EGF) into signaling events promoting cell growth and proliferation in many mammalian cell types. In a simplified model, the presence of mitogens and growth factors trigger the activation of canonical receptor tyrosine kinases such as EGFR leading to their dimerization and subsequent activation of the small GTPase Ras.[6] This then leads to a series of phosphorylation events downstream in the MAPK cascade (Raf-MEK-ERK) ultimately resulting in the phosphorylation and activation of ERK. The phosphorylation of ERK results in an activation of its kinase activity and leads to phosphorylation of its many downstream targets involved in regulation of cell proliferation. In most cells, some form of sustained ERK activity is required for cells to activate genes that induce cell cycle entry and suppress negative regulators of the cell cycle. Two such important targets include Cyclin D complexes with Cdk4 and Cdk6 (Cdk4/6) which are both phosphorylated by ERK.[7] The transition from G1 to S phase is coordinated by the activity of Cyclin D-Cdk4/6, which increases during late G1 phase as cells prepare to enter S-phase in response to mitogens. Cdk4/6 activation contributes to hyper-phosphorylation and the subsequent destabilization of retinoblastoma protein (Rb).[7] Hypo-phosphorylated Rb, is normally bound to transcription factor E2F in early G1 and inhibits its transcriptional activity, preventing expression of S-phase entry genes including Cyclin E, Cyclin A2 and Emi1.[6] ERK1/2 activation downstream of mitogen induced Ras signaling is necessary and sufficient to remove this cell cycle block and allow cells to progress to S-phase in most mammalian cells.

Downstream feedback control and generation of a bistable G1/S switch
The restriction point (R-point) marks the critical event when a mammalian cell commits to proliferation and becomes independent of growth stimulation. It is fundamental for normal differentiation and tissue homeostasis, and seems to be dysregulated in virtually all cancers. Although the R-point has been linked to various activities involved in the regulation of G1–S transition of the mammalian cell cycle, the underlying mechanism remains unclear. Using single-cell measurements, Yao et al., shows that the Rb–E2F pathway functions as a bistable switch to convert graded serum inputs into all-or-none E2F responses.[8]
Growth and mitogen signals are transmitted downstream of the ERK pathway are incorporated into multiple positive feedback loops to generate a bistable switch at the level of E2F activation.[8] This occurs due to three main interactions during late G1 phase. The first is a result of mitogen stimulation though the ERK leading to the expression of the transcription factor Myc, which is a direct activator of E2F.[7] The second pathway is a result of ERK activation leading to the accumulation of active complexes of Cyclin D and Cdk4/6 which destabilize Rb via phosphorylation and further serve to activate E2F and promote expression of its targets. Finally, these interactions are all reinforced by an additional positive feedback loop by E2F on itself, as its own expression leads to production of the active complex of Cyclin E and CDK2, which further serves to lock in a cell's decision to enter S-phase. As a result, when serum concentration is increased in a gradual manner, most mammalian cells respond in a switch-like manner in entering S-phase. This mitogen stimulated, bistable E2F switch is exhibits hysteresis, as cells are inhibited from returning to G1 even after mitogen withdrawal post E2F activation.[9]
Dynamic signal processing by the ERK pathway
The EGFR-ERK/MARK (epidermal growth factor receptor extracellular-regulated kinase/mitogen-activated protein kinase) pathway stimulated by EGF is critical for cellular proliferation, but the temporal separation between signal and response obscures the signal-response relationship in previous research.In 2013, Albeck et al.[10] provided key experimental evidence to fill this gap of knowledge. They measured signal strength and dynamics with steady-state EGF stimulation, in which the signaling and output can be easily related. They further mapped the signal-response relationship across the pathway’s full dynamic range. Using high-content immunofluorescence (HCIF) detection of phosphorylated ERK (pERK) and live cell FRET biosensors, they monitored downstream output of the ERK pathway in both live cells and fixed cells. To further link the quantitative characteristics of ERK signaling to proliferation rates, they established a series of steady-state conditions using a range of EGF concentrations by applying EGF with different concentrations.
Single cell imaging experiments have shown ERK to be activated in stochastic bursts in the presence of EGF. Furthermore, the pathway has been shown to encode the strength of signaling inputs though frequency modulated pulses of its activity. Using live cell FRET biosensors, cells induced with different concentrations of EGF illicit activity bursts of different frequency, where higher levels of EGF resulted in more frequent bursts of ERK activity. To figure out how S phase entry can be affected by sporadic pulses of ERK activity at low EGF concentrations, they used MCF-10A cells co-expressing EKAR-EV and RFP-geminin and identified the pulses of ERK activity with the scoring and then align this ERK activity profiles with time of GFP-geminin induction. They found that longer periods of ERK activity stimulate S phase entry, as suggested by increased pulse length. To understand the dynamics of EGFR-ERK pathway, specifically how is the frequency and amplitude modulated, they applied the EGFR inhibitor gefitinib or the highly selective MAPK/ERK kinase (MEK) inhibitor PD0325901 (PD). Two inhibitors yield actually a little bit different result: gefitinib, at intermediate concentration, would induce pulsatory behavior and also bimodal shift, which is not observed with PD. They further combine EGF and PD together and draw the conclusion that the frequency of ERK activities is modulated by quantitative variation while the amplitude is modulated by MEK activity’s change. Lastly they turned to Fra-1, one of downstream effectors of ERK pathway, as it’s technically challenging to estimate ERK activities directly. To understand how the integrated ERK pathway output (which should be independent of either frequency or amplitude) affect the proliferation rate, they used the combination of a wide range of EGF and PD concentrations and find that there’s actually an inverted “L” shape single curvilinear relationship, which suggests that at low levels of ERK pathway output, small changes in signal intensity correspond to large changes in proliferative rate, while large changes in signal intensity near the high end of the dynamic range have little impact on proliferation. The fluctuation of ERK signaling highlights potential issues with current therapeutic approaches, providing new perspective in terms of thinking about drug targeting in the ERK pathway in cancer.

Integration of mitogen and stress signals in proliferation
Recent live cell imaging experiments in MCF10A and MCF7 cells have shown that a combination of mitogen signaling though ERK and stress signals through activation of p53 in mother cells contributes to the likelihood of whether newly formed daughter cells will immediately re-enter the cell cycle or enter quiescence (G0) preceding mitosis.[11] Rather than daughter cells starting with no key signaling proteins after division, mitogen/ERK induced Cyclin D1 mRNA and DNA damage induced p53 protein, both long lived factors in cells, can be stably inherited from mother cells after cell division. The levels of these regulators vary from cell to cell after mitosis and stoichiometry between them strongly influences cell cycle commitment though activation of Cdk2. Chemical perturbations using inhibitors of ERK signaling or inducers p53 signaling in mother cells suggest daughter cells with high levels of p53 protein and low levels of Cyclin D1 transcripts were shown to primarily enter G0 whereas cells with high Cyclin D1 and low levels of p53 are most likely to reenter the cell cycle. These results illustrate a form of encoded molecular memory though the history of mitogen signaling through ERK and stress response though p53.[12][13]
Clinical significance
Uncontrolled growth is a necessary step for the development of all cancers.[14] In many cancers (e.g. melanoma), a defect in the MAP/ERK pathway leads to that uncontrolled growth. Many compounds can inhibit steps in the MAP/ERK pathway, and therefore are potential drugs for treating cancer,[15][16][17][18][19] e.g., Hodgkin disease.[20]
The first drug licensed to act on this pathway is sorafenib — a Raf kinase inhibitor.
Other Raf inhibitors: SB590885, PLX4720, XL281, RAF265, encorafenib, dabrafenib, vemurafenib.[19]
Some MEK inhibitors: cobimetinib, CI-1040, PD0325901, Binimetinib (MEK162), selumetinib,[19] Trametinib(GSK1120212)[21] It has been found that acupoint-moxibustion has a role in relieving alcohol-induced gastric mucosal injury in a mouse model, which may be closely associated with its effects in up-regulating activities of the epidermal growth factor/ERK signal transduction pathway.[22]
RAF-ERK pathway is also involved in the pathophysiology of Noonan's Syndrome, a polymalformative disease, where Simvastatin was proposed as a way to improve CNS-cognitive symptoms of the disorder.
Protein microarray analysis can be used to detect subtle changes in protein activity in signaling pathways.[23] The developmental syndromes caused by germline mutations in genes that alter the RAS components of the MAP/ERK signal transduction pathway are called RASopathies.
See also
- Janus kinase
- Phosphatase
- Signal transducing adaptor protein
- G protein-coupled receptor
References
- ↑ Orton RJ, Sturm OE, Vyshemirsky V, Calder M, Gilbert DR, Kolch W (Dec 2005). "Computational modelling of the receptor-tyrosine-kinase-activated MAPK pathway". The Biochemical Journal. 392 (Pt 2): 249–61. doi:10.1042/BJ20050908. PMC 1316260. PMID 16293107.
- ↑ Schulze WX, Deng L, Mann M (2005). "Phosphotyrosine interactome of the ErbB-receptor kinase family". Molecular Systems Biology. 1 (1): 2005.0008. doi:10.1038/msb4100012. PMC 1681463. PMID 16729043.
- ↑ Zarich N, Oliva JL, Martínez N, et al. (Aug 2006). "Grb2 is a negative modulator of the intrinsic Ras-GEF activity of hSos1". Molecular Biology of the Cell. 17 (8): 3591–7. doi:10.1091/mbc.E05-12-1104. PMC 1525251. PMID 16760435.
- 1 2 Avruch J, Khokhlatchev A, Kyriakis JM, et al. (2001). "Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade". Recent Progress in Hormone Research. 56 (1): 127–55. doi:10.1210/rp.56.1.127. PMID 11237210.
- ↑ Pende M, Um SH, Mieulet V, et al. (Apr 2004). "S6K1,(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5'-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway". Molecular and Cellular Biology. 24 (8): 3112–24. doi:10.1128/MCB.24.8.3112-3124.2004. PMC 381608. PMID 15060135.
- 1 2 Meloche, et al.. “The ERK1/2 Mitogen-Activated Protein Kinase Pathway as a Master Regulator of the G1- to S-Phase Transition.” Oncogene, vol. 26, no. 22, 2007, pp. 3227–3239., doi:10.1038/sj.onc.1210414.
- 1 2 3 Chambard, Jean-Claude, et al. “ERK Implication in Cell Cycle Regulation.” Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1773, no. 8, 2007, pp. 1299–1310., doi:10.1016/j.bbamcr.2006.11.010.
- 1 2 4. Yao, Guang, et al. “A Bistable Rb-E2F Switch Underlies the Restriction Point.” Nature Cell Biology, vol. 10, no. 4, 2008, pp. 476–482., doi:10.1038/ncb1711.
- ↑ Yao, Guang, et al. “A Bistable Rb-E2F Switch Underlies the Restriction Point.” Nature Cell Biology, vol. 10, no. 4, 2008, pp. 476–482., doi:10.1038/ncb1711.
- ↑ Albeck, John A G., et al. “Frequency-Modulated Pulses of ERK Activity Transmit Quantitative Proliferation Signals.” Molecular Cell, vol. 49, no. 2, 2013, pp. 249–261., doi:10.1016/j.molcel.2012.11.002.
- ↑ 8. Yang, Hee Won, et al. “Competing Memories of Mitogen and p53 Signalling Control Cell-Cycle Entry.” Nature, vol. 549, no. 7672, June 2017, pp. 404–408., doi:10.1038/nature23880.
- ↑ Yang, Hee Won, et al. “Competing Memories of Mitogen and p53 Signalling Control Cell-Cycle Entry.” Nature, vol. 549, no. 7672, June 2017, pp. 404–408., doi:10.1038/nature23880.
- ↑ Kedziora, Katarzyna M., and Jeremy E. Purvis. “Cell Biology: The Persistence of Memory.”Nature, vol. 549, no. 7672, June 2017, pp. 343–344., doi:10.1038/nature23549.
- ↑ Downward J (2003). "Targeting RAS signalling pathways in cancer therapy". Nature Reviews Cancer. 3 (1): 11–22. doi:10.1038/nrc969. PMID 12509763. S2CID 43074411.
- ↑ Hilger RA, Scheulen ME, Strumberg D (December 2002). "The Ras-Raf-MEK-ERK pathway in the treatment of cancer" (PDF). Onkologie. 25 (6): 511–8. doi:10.1159/000068621. PMID 12566895. S2CID 26673969. Archived from the original on 2012-10-06.
- ↑ Sebolt-Leopold JS (June 2008). "Advances in the development of cancer therapeutics directed against the RAS-mitogen-activated protein kinase pathway". Clin. Cancer Res. 14 (12): 3651–6. doi:10.1158/1078-0432.CCR-08-0333. PMID 18559577. Archived from the original on 2011-09-06.
- ↑ Hoshino R, Chatani Y, Yamori T, et al. (January 1999). "Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors". Oncogene. 18 (3): 813–22. doi:10.1038/sj.onc.1202367. PMID 9989833.
- ↑ McCubrey JA, Steelman LS, Chappell WH, et al. (August 2007). "Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance". Biochim. Biophys. Acta. 1773 (8): 1263–84. doi:10.1016/j.bbamcr.2006.10.001. PMC 2696318. PMID 17126425.
- 1 2 3 Kwong-Kwok Wong (2009). "Recent Developments in Anti-Cancer Agents Targeting the Ras/Raf/ MEK/ERK Pathway" (PDF). Archived from the original (PDF) on 2010-06-16.
{{cite journal}}: Cite journal requires|journal=(help) - ↑ Zheng B, Fiumara P, Li YV, et al. (August 2003). "MEK/ERK pathway is aberrantly active in Hodgkin disease: a signaling pathway shared by CD30, CD40, and RANK that regulates cell proliferation and survival". Blood. 102 (3): 1019–27. doi:10.1182/blood-2002-11-3507. PMID 12689928.
- ↑ "Archived copy". Archived from the original on 2012-07-08. Retrieved 2010-11-26.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ Zhang H, Guo H, Zhang YC, Liu M, Ai K, Su YM, Li MH, Li TL (2014). "[Effect of moxibustion intervention on expression of gastric epidermal growth factor receptor and extracellular signal regulated kinase 1/2 expression in rats with gastric ulcer]". Zhen Ci Yan Jiu. 39 (5): 351–7. PMID 25518106.
- ↑ Calvert VS, Tang Y, Boveia V, Wulfkuhle J, Schutz-Geschwender A, Oliver DM, Liotta LA, Petricoin EF (2004). "Development of Multiplexed Protein Profiling and Detection Using Near Infrared Detection of Reverse-Phase Protein Microarrays". Clinical Proteomics Journal. 1 (1): 81–89. doi:10.1385/CP:1:1:081.
External links
- MAP Kinase Resource .
- Kyoto Encyclopedia of Genes and Genomes — MAPK pathway
- MAP+Kinase+Signaling+System at the US National Library of Medicine Medical Subject Headings (MeSH)