Tumor suppressor gene
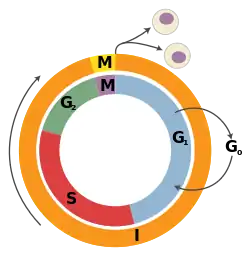
A tumor suppressor gene (TSG), or anti-oncogene, is a gene that regulates a cell during cell division and replication.[1] If the cell grows uncontrollably, it will result in cancer. When a tumor suppressor gene is mutated, it results in a loss or reduction in its function. In combination with other genetic mutations, this could allow the cell to grow abnormally. The loss of function for these genes may be even more significant in the development of human cancers, compared to the activation of oncogenes.[2]
TSGs can be grouped into the following categories: caretaker genes, gatekeeper genes, and more recently landscaper genes. Caretaker genes ensure stability of the genome via DNA repair and subsequently when mutated allow mutations to accumulate.[3] Meanwhile, gatekeeper genes directly regulate cell growth by either inhibiting cell cycle progression or inducing apoptosis.[3] Lastly landscaper genes regulate growth by contributing to the surrounding environment, when mutated can cause an environment that promotes unregulated proliferation.[4] The classification schemes are evolving as medical advances are being made from fields including molecular biology, genetics, and epigenetics.
History
The discovery of oncogenes and their ability to deregulate cellular processes related to cell proliferation and development appeared first in the literature as opposed to the idea of tumor suppressor genes.[5] However, the idea of genetic mutation leading to increased tumor growth gave way to another possible genetic idea of genes playing a role in decreasing cellular growth and development of cells. This idea was not solidified until experiments by Henry Harris were conducted with somatic cell hybridization in 1969.[6]
Within Harris's experiments, tumor cells were fused with normal somatic cells to make hybrid cells. Each cell had chromosomes from both parents and upon growth, a majority of these hybrid cells did not have the capability of developing tumors within animals.[6] The suppression of tumorigenicity in these hybrid cells prompted researchers to hypothesize that genes within the normal somatic cell had inhibitory actions to stop tumor growth.[6] This initial hypothesis eventually lead to the discovery of the first classic tumor suppressor gene by Alfred Knudson, known as the Rb gene, which codes for the retinoblastoma tumor suppressor protein.[5]
Alfred Knudson, a pediatrician and cancer geneticist, proposed that in order to develop retinoblastoma, two allelic mutations are required to lose functional copies of both the Rb genes to lead to tumorigenicity.[6] Knudson observed that retinoblastoma often developed early in life for younger patients in both eyes, while in some rarer cases retinoblastoma would develop later in life and only be unilateral.[5] This unique development pattern allowed Knudson and several other scientific groups in 1971 to correctly hypothesize that the early development of retinoblastoma was caused by inheritance of one loss of function mutation to an RB germ-line gene followed by a later de novo mutation on its functional Rb gene allele. The more sporadic occurrence of unilateral development of retinoblastoma was hypothesized to develop much later in life due to two de novo mutations that were needed to fully lose tumor suppressor properties.[5] This finding formed the basis of the two-hit hypothesis. In order to verify that the loss of function of tumor suppressor genes causes increased tumorigenicity, interstitial deletion experiments on chromosome 13q14 were conducted to observe the effect of deleting the loci for the Rb gene. This deletion caused increased tumor growth in retinoblastoma, suggesting that loss or inactivation of a tumor suppressor gene can increase tumorigenicity.[6]
Two-hit hypothesis
Unlike oncogenes, tumor suppressor genes generally follow the two-hit hypothesis, which states both alleles that code for a particular protein must be affected before an effect is manifested.[7] If only one allele for the gene is damaged, the other can still produce enough of the correct protein to retain the appropriate function. In other words, mutant tumor suppressor alleles are usually recessive, whereas mutant oncogene alleles are typically dominant.

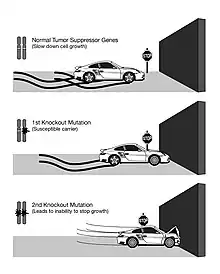
Proposed by A.G. Knudson for cases of retinoblastoma.[7] He observed that 40% of U.S cases were caused by a mutation in the germ-line. However, affected parents could have children without the disease, but the unaffected children became parents of children with retinoblastoma.[8] This indicates that one could inherit a mutated germ-line but not display the disease. Knudson observed that the age of onset of retinoblastoma followed 2nd order kinetics, implying that two independent genetic events were necessary. He recognized that this was consistent with a recessive mutation involving a single gene, but requiring bi-allelic mutation. Hereditary cases involve an inherited mutation and a single mutation in the normal allele.[8] Non-hereditary retinoblastoma involves two mutations, one on each allele.[8] Knudson also noted that hereditary cases often developed bilateral tumors and would develop them earlier in life, compared to non-hereditary cases where individuals were only affected by a single tumor.[8]
There are exceptions to the two-hit rule for tumor suppressors, such as certain mutations in the p53 gene product. p53 mutations can function as a dominant negative, meaning that a mutated p53 protein can prevent the function of the natural protein produced from the non-mutated allele.[9] Other tumor-suppressor genes that do not follow the two-hit rule are those that exhibit haploinsufficiency, including PTCH in medulloblastoma and NF1 in neurofibroma. Another example is p27, a cell-cycle inhibitor, that when one allele is mutated causes increased carcinogen susceptibility.[10]
Functions
The proteins encoded by most tumor suppressor genes inhibit cell proliferation or survival. Inactivation of tumor suppressor genes therefore leads to tumor development by eliminating negative regulatory proteins. In most cases, tumor suppressor proteins inhibit the same cell regulatory pathways that are stimulated by the products of oncogenes.[11] While tumor suppressor genes have the same main function, they have various mechanisms of action, that their transcribed products perform, which include the following:[12]
- Intracellular proteins, that control gene expression of a specific stage of the cell cycle. If these genes are not expressed, the cell cycle does not continue, effectively inhibiting cell division. (e.g., pRB and p16)[13]
- Receptors or signal transducers for secreted hormones or developmental signals that inhibit cell proliferation (e.g., transforming growth factor (TGF)-β and adenomatous polyposis coli (APC)).[14]
- Checkpoint-control proteins that trigger cell cycle arrest in response to DNA damage or chromosomal defects (e.g., breast cancer type 1 susceptibility protein (BRCA1), p16, and p14).[15]
- Proteins that induce apoptosis. If damage cannot be repaired, the cell initiates programmed cell death to remove the threat it poses to the organism as a whole. (e.g., p53).[16]
- Cell adhesion. Some proteins involved in cell adhesion prevent tumor cells from dispersing, block loss of contact inhibition, and inhibit metastasis. These proteins are known as metastasis suppressors. (e.g., CADM1)[17][18]
- Proteins involved in repairing mistakes in DNA. Caretaker genes encode proteins that function in repairing mutations in the genome, preventing cells from replicating with mutations. Furthermore, increased mutation rate from decreased DNA repair leads to increased inactivation of other tumor suppressors and activation of oncogenes.[19] (e.g., p53 and DNA mismatch repair protein 2 (MSH2)).[20]
- Certain genes can also act as tumor suppressors and oncogenes. Dubbed Proto-oncogenes with Tumor suppressor function, these genes act as “double agents” that both positively and negatively regulate transcription. (e.g., NOTCH receptors, TP53 and FAS).[21]
Epigenetic influences
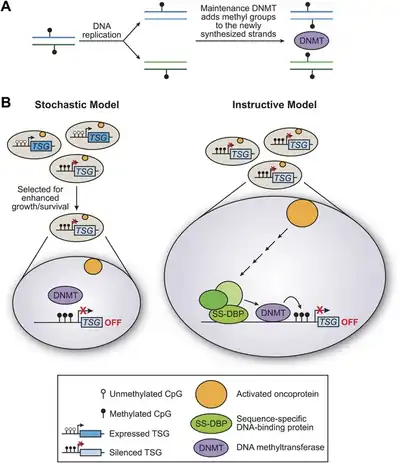
Expression of genes, including tumor suppressors, can be altered through biochemical alterations known as DNA methylation.[23] Methylation is an example of epigenetic modifications, which commonly regulate expression in mammalian genes. The addition of a methyl group to either histone tails or directly on DNA causes the nucleosome to pack tightly together restricting the transcription of any genes in this region. This process not only has the capabilities to inhibit gene expression, it can also increase the chance of mutations. Stephen Baylin observed that if promoter regions experience a phenomenon known as hypermethylation, it could result in later transcriptional errors, tumor suppressor gene silencing, protein misfolding, and eventually cancer growth. Baylin et al. found methylation inhibitors known as azacitidine and decitabine. These compounds can actually help prevent cancer growth by inducing re-expression of previously silenced genes, arresting the cell cycle of the tumor cell and forcing it into apoptosis.[24]
There are further clinical trials under current investigation regarding treatments for hypermethylation as well as alternate tumor suppression therapies that include prevention of tissue hyperplasia, tumor development, or metastatic spread of tumors.[25] The team working with Wajed have investigated neoplastic tissue methylation in order to one day identify early treatment options for gene modification that can silence the tumor suppressor gene.[26] In addition to DNA methylation, other epigenetic modifications like histone deacetylation or chromatin-binding proteins can prevent DNA polymerase from effectively transcribing desired sequences, such as ones containing tumor suppressor genes.
Clinical significance
Gene therapy is used to reinstate the function of a mutated or deleted gene type. When tumor suppressor genes are altered in a way that results in less or no expression, several severe problems can arise for the host. This is why tumor suppressor genes have commonly been studied and used for gene therapy. The two main approaches used currently to introduce genetic material into cells are viral and non-viral delivery methods.[26]
Viral methods
The viral method of transferring genetic material harnesses the power of viruses.[26] By using viruses that are durable to genetic material alterations, viral methods of gene therapy for tumor suppressor genes have shown to be successful.[27] In this method, vectors from viruses are used. The two most commonly used vectors are adenoviral vectors and adeno-associated vectors. In vitro genetic manipulation of these types of vectors is easy and in vivo application is relatively safe compared to other vectors.[26][28] Before the vectors are inserted into the tumors of the host, they are prepared by having the parts of their genome that control replication either mutated or deleted. This makes them safer for insertion. Then, the desired genetic material is inserted and ligated to the vector.[27] In the case with tumor suppressor genes, genetic material which encodes p53 has been used successfully, which after application, has shown reduction in tumor growth or proliferation.[28][29]
Non-viral methods
The non-viral method of transferring genetic material is used less often than the viral method.[26][28] However, the non-viral method is a more cost-effective, safer, available method of gene delivery not to mention that non-viral methods have shown to induce fewer host immune responses and possess no restrictions on size or length of the transferable genetic material.[26] Non-viral gene therapy uses either chemical or physical methods to introduce genetic material to the desired cells.[26][28] The chemical methods are used primarily for tumor suppressor gene introduction and are divided into two categories which are naked plasmid or liposome-coated plasmids.[28] The naked plasmid strategy has garnered interest because of its easy to use methods.[26] Direct injection into the muscles allows for the plasmid to be taken up into the cell of possible tumors where the genetic material of the plasmid can be incorporated into the genetic material of the tumor cells and revert any previous damage done to tumor suppressor genes.[26][28] The liposome-coated plasmid method has recently also been of interest since they produce relatively low host immune response and are efficient with cellular targeting.[28] The positively charged capsule in which the genetic material is packaged helps with electrostatic attraction to the negatively charged membranes of the cells as well as the negatively charged DNA of the tumor cells.[26][28] In this way, non-viral methods of gene therapy are highly effective in restoring tumor suppressor gene function to tumor cells that have either partially or entirely lost this function.
Limitations
The viral and non-viral gene therapies mentioned above are commonly used but each has some limitations which must be considered. The most important limitation these methods have is the efficacy at which the adenoviral and adeno-associated vectors, naked plasmids, or liposome-coated plasmids are taken in by the host’s tumor cells. If proper uptake by the host’s tumor cells is not achieved, re-insertion introduces problems such as the host’s immune system recognizing these vectors or plasmids and destroying them which impairs the overall effectiveness of the gene therapy treatment further.[29]
Examples
| Gene | Original Function | Two-Hit? | Associated Carcinomas |
|---|---|---|---|
| Rb | DNA Replication, cell division and death | Yes | Retinoblastoma[5] |
| p53 | Apoptosis | No | Half of all known malignancies[5] |
| VHL | Cell division, death, and differentiation | Yes | Kidney Cancer[26] |
| APC | DNA damage, cell division, migration, adhesion, death | Yes | Colorectal Cancer[26] |
| BRCA2 | Cell division and death, and repair of double-stranded DNA breaks | Yes | Breast/Ovarian Cancer[5] |
| NF1 | Cell differentiation, division, development, RAS signal transduction | No | Nerve tumors, Neuroblastoma[26] |
| PTCH | Hedgehog signaling | No | Medulloblastoma, Basal Cell Carcinoma[5] |
- Retinoblastoma protein (pRb). pRb was the first tumor-suppressor protein discovered in human retinoblastoma; however, recent evidence has also implicated pRb as a tumor-survival factor. RB1 gene is a gatekeeper gene that blocks cell proliferation, regulates cell division and cell death.[8] Specifically pRb prevents the cell cycle progression from G1 phase into the S phase by binding to E2F and repressing the necessary gene transcription.[30] This prevents the cell from replicating its DNA if there is damage.
- p53. TP53, a caretaker gene, encodes the protein p53, which is nicknamed "the guardian of the genome". p53 has many different functions in the cell including DNA repair, inducing apoptosis, transcription, and regulating the cell cycle.[31] Mutated p53 is involved in many human cancers, of the 6.5 million cancer diagnoses each year about 37% are connected to p53 mutations.[31] This makes it a popular target for new cancer therapies. Homozygous loss of p53 is found in 65% of colon cancers, 30–50% of breast cancers, and 50% of lung cancers. Mutated p53 is also involved in the pathophysiology of leukemias, lymphomas, sarcomas, and neurogenic tumors. Abnormalities of the p53 gene can be inherited in Li-Fraumeni syndrome (LFS), which increases the risk of developing various types of cancers.
- BCL2. BCL2 is a family of proteins that are involved in either inducing or inhibiting apoptosis.[32] The main function is involved in maintaining the composition of the mitochondria membrane, and preventing cytochrome c release into the cytosol.[32] When cytochrome c is released from the mitochondria it starts a signaling cascade to begin apoptosis.[33]
- SWI/SNF. SWI/SNF is a chromatin remodeling complex, which is lost in about 20% of tumors.[34] The complex consists of 10-15 subunits encoded by 20 different genes.[34] Mutations in the individual complexes can lead to misfolding, which compromises the ability of the complex to work together as a whole. SWI/SNF has the ability move nucleosomes, which condenses DNA, allowing for transcription or block transcription from occurring for certain genes.[34] Mutating this ability could cause genes to be turned on or off at the wrong times.
As the cost of DNA sequencing continues to diminish, more cancers can be sequenced. This allows for the discovery of novel tumor suppressors and can give insight on how to treat and cure different cancers in the future. Other examples of tumor suppressors include pVHL, APC, CD95, ST5, YPEL3, ST7, and ST14, p16, BRCA2.[35]
See also
- Anticancer gene
- Metastasis suppressor
- Adenomatosis polyposis coli
- Oncogene
- Cancer
- DNA repair
- Signal transduction
- Von Hippel Lindau Binding protein 1
- BRCA1
- p53
References
- ↑ "Oncogenes and tumor suppressor genes | American Cancer Society". www.cancer.org. Archived from the original on 2021-03-18. Retrieved 2019-09-26.
- ↑ Weinberg, Robert A (2014). "The Biology of Cancer." Garland Science, page 231.
- 1 2 "Glossary of Cancer Genetics (side-frame)". www.cancerindex.org. Archived from the original on 2020-10-07. Retrieved 2019-11-19.
- ↑ "Cancer Genetics - CuboCube". www.cubocube.com. Archived from the original on 2020-10-12. Retrieved 2019-11-19.
- 1 2 3 4 5 6 7 8 Sherr, Charles J. (2004). "Principles of Tumor Suppression". Cell. 116 (2): 235–246. doi:10.1016/S0092-8674(03)01075-4. PMID 14744434. S2CID 18712326.
- 1 2 3 4 5 Cooper, G. M. (2000). Tumor Suppressor Genes. The Cell: A Molecular Approach. 2nd Edition. https://www.ncbi.nlm.nih.gov/books/NBK9894/ Archived 2021-01-19 at the Wayback Machine
- 1 2 Knudson AG (April 1971). "Mutation and cancer: statistical study of retinoblastoma". Proceedings of the National Academy of Sciences of the United States of America. 68 (4): 820–823. Bibcode:1971PNAS...68..820K. doi:10.1073/pnas.68.4.820. PMC 389051. PMID 5279523.
- 1 2 3 4 5 "Tumor Suppressor (TS) Genes and the Two-Hit Hypothesis | Learn Science at Scitable". www.nature.com. Archived from the original on 2017-05-30. Retrieved 2019-10-06.
- ↑ Baker SJ, Markowitz S, Fearon ER, Willson JK, Vogelstein B (August 1990). "Suppression of human colorectal carcinoma cell growth by wild-type p53". Science. 249 (4971): 912–915. Bibcode:1990Sci...249..912B. doi:10.1126/science.2144057. PMID 2144057.
- ↑ Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ (November 1998). "The murine gene p27Kip1 is haplo-insufficient for tumour suppression". Nature. 396 (6707): 177–180. Bibcode:1998Natur.396..177F. doi:10.1038/24179. PMC 5395202. PMID 9823898.
- ↑ Cooper GM (2000). "Tumor Suppressor Genes". The Cell: A Molecular Approach (2nd ed.). Sunderland (MA): Sinauer Associates. Archived from the original on 2021-01-19. Retrieved 2023-08-18.
- ↑ Wang LH, Wu CF, Rajasekaran N, Shin YK (2018). "Loss of Tumor Suppressor Gene Function in Human Cancer: An Overview". Cellular Physiology and Biochemistry. 51 (6): 2647–2693. doi:10.1159/000495956. PMID 30562755.
- ↑ Leiderman YI, Kiss S, Mukai S (2007). "Molecular genetics of RB1--the retinoblastoma gene". Seminars in Ophthalmology. 22 (4): 247–254. doi:10.1080/08820530701745165. PMID 18097988. S2CID 42925807.
- ↑ Smith AL, Robin TP, Ford HL (September 2012). "Molecular pathways: targeting the TGF-β pathway for cancer therapy". Clinical Cancer Research. 18 (17): 4514–4521. doi:10.1158/1078-0432.CCR-11-3224. PMID 22711703.
- ↑ Savage KI, Harkin DP (February 2015). "BRCA1, a 'complex' protein involved in the maintenance of genomic stability". The FEBS Journal. 282 (4): 630–646. doi:10.1111/febs.13150. PMID 25400280.
- ↑ Nayak SK, Panesar PS, Kumar H (2009). "p53-Induced apoptosis and inhibitors of p53". Current Medicinal Chemistry. 16 (21): 2627–2640. doi:10.2174/092986709788681976. PMID 19601800.
- ↑ Yoshida BA, Sokoloff MM, Welch DR, Rinker-Schaeffer CW (November 2000). "Metastasis-suppressor genes: a review and perspective on an emerging field". Journal of the National Cancer Institute. 92 (21): 1717–1730. doi:10.1093/jnci/92.21.1717. PMID 11058615.
- ↑ Hirohashi S, Kanai Y (July 2003). "Cell adhesion system and human cancer morphogenesis". Cancer Science. 94 (7): 575–581. doi:10.1111/j.1349-7006.2003.tb01485.x. PMID 12841864. S2CID 22154824.
- ↑ Markowitz S (November 2000). "DNA repair defects inactivate tumor suppressor genes and induce hereditary and sporadic colon cancers". Journal of Clinical Oncology. 18 (21 Suppl): 75S–80S. PMID 11060332.
- ↑ Rahman N, Scott RH (April 2007). "Cancer genes associated with phenotypes in monoallelic and biallelic mutation carriers: new lessons from old players". Human Molecular Genetics. 16 Spec No 1 (R1): R60–R66. doi:10.1093/hmg/ddm026. PMID 17613548.
- ↑ Shen L, Shi Q, Wang W (March 2018). "Double agents: genes with both oncogenic and tumor-suppressor functions". Oncogenesis. 7 (3): 25. doi:10.1038/s41389-018-0034-x. PMC 5852963. PMID 29540752.
- ↑ Struhl, Kevin (12 March 2014). "Is DNA methylation of tumour suppressor genes epigenetic?". eLife. 3: e02475. doi:10.7554/eLife.02475. ISSN 2050-084X. Retrieved 22 October 2023.
- ↑ Wajed SA, Laird PW, DeMeester TR (July 2001). "DNA methylation: an alternative pathway to cancer". Annals of Surgery. 234 (1): 10–20. doi:10.1097/00000658-200107000-00003. PMC 1421942. PMID 11420478.
- ↑ Baylin SB (December 2005). "DNA methylation and gene silencing in cancer". Nature Clinical Practice. Oncology. 2 (Suppl 1): S4-11. doi:10.1038/ncponc0354. PMID 16341240. S2CID 19361179.
- ↑ Delbridge AR, Valente LJ, Strasser A (November 2012). "The role of the apoptotic machinery in tumor suppression". Cold Spring Harbor Perspectives in Biology. 4 (11): a008789. doi:10.1101/cshperspect.a008789. PMC 3536334. PMID 23125015.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 "Tumor Suppressor (TS) Genes and the Two-Hit Hypothesis | Learn Science at Scitable". www.nature.com. Archived from the original on 2017-05-30. Retrieved 2020-10-27.
- 1 2 Nayerossadat N, Maedeh T, Ali PA (2012-07-06). "Viral and nonviral delivery systems for gene delivery". Advanced Biomedical Research. 1: 27. doi:10.4103/2277-9175.98152. PMC 3507026. PMID 23210086.
- 1 2 3 4 5 6 7 8 Guo XE, Ngo B, Modrek AS, Lee WH (January 2014). "Targeting tumor suppressor networks for cancer therapeutics". Current Drug Targets. 15 (1): 2–16. doi:10.2174/1389450114666140106095151. PMC 4032821. PMID 24387338.
- 1 2 Morris LG, Chan TA (May 2015). "Therapeutic targeting of tumor suppressor genes". Cancer. 121 (9): 1357–1368. doi:10.1002/cncr.29140. PMC 4526158. PMID 25557041.
- ↑ "RETINOBLASTOMA: Protein". dpuadweb.depauw.edu. Archived from the original on 2019-09-08. Retrieved 2019-11-21.
- 1 2 Harris CC (October 1996). "Structure and function of the p53 tumor suppressor gene: clues for rational cancer therapeutic strategies". Journal of the National Cancer Institute. 88 (20): 1442–1455. doi:10.1093/jnci/88.20.1442. PMID 8841019.
- 1 2 "BCL2 (B-Cell Leukemia/Lymphoma 2)". atlasgeneticsoncology.org. Archived from the original on 2021-06-14. Retrieved 2019-11-21.
- ↑ Goodsell DS (2004-04-01). "The molecular perspective: cytochrome C and apoptosis". The Oncologist. 9 (2): 226–227. doi:10.1634/theoncologist.9-2-226. PMID 15047927.
- 1 2 3 Shain AH, Pollack JR (2013). "The spectrum of SWI/SNF mutations, ubiquitous in human cancers". PLOS ONE. 8 (1): e55119. Bibcode:2013PLoSO...855119S. doi:10.1371/journal.pone.0055119. PMC 3552954. PMID 23355908.
- ↑ "TUMOUR SUPPRESSOR GENES IN CANCER". www.letstalkacademy.com. Archived from the original on 2021-04-19. Retrieved 2019-11-21.
External links
- TCF21 gene discovery at Ohio State University Archived 2009-02-04 at the Wayback Machine
- Drosophila Oncogenes and Tumor Suppressors - The Interactive Fly Archived 2013-11-09 at the Wayback Machine
- Tumor Suppressor Gene Database, published in 2012 Archived 2014-01-09 at the Wayback Machine