11p deletion syndrome
| 11p deletion syndrome | |
|---|---|
| Other names: WARG syndrome, WAGR complex, Wilms tumour-aniridia syndrome, aniridia-Wilms tumour syndrome, Miller syndrome[1] | |
| Video explanation | |
| Specialty | Medical genetics |
| Symptoms | Wilms tumor, absence of the colored part of the eye, undescended testicles, intellectual disability[1] |
| Complications | Cataracts, childhood obesity, pancreatitis, kidney failure[1] |
| Usual onset | Present at birth[1] |
| Causes | Genetic deletion from the short arm of chromosome 11[1] |
| Diagnostic method | Suspected based on examination, confirmed by karyotyping[2] |
| Differential diagnosis | Drash syndrome, Frasier syndrome, Potocki-Shaffer syndrome[2] |
| Treatment | Determined by the symptoms[2] |
| Prognosis | Decrease life expectancy[3] |
| Frequency | 1 in 750,000[1] |
11p deletion syndrome, previously known as WAGR syndrome, is genetic disorder in which people are at risk of Wilms tumor (a type of kidney cancer), aniridia (absence of the colored part of the eye), genitourinary abnormalities such as undescended testicles, and mental retardation.[1] Other symptoms may include light sensitivity, cataracts, childhood obesity, pancreatitis, and kidney failure.[1] Intellectual disabilities are generally in the mild to moderate range.[2]
The cause is the deletion of genetic material on the short arm of chromosome 11.[1] Most cases occur spontaneously during early development, though rarely it may be inherited from a parent.[1] What is missing is a group of genes, including PAX6 and WT1, that sit next to each other.[1] If the BDNF gene is also involved obesity occurs.[1] Diagnosis is generally suspected based on examination at birth and confirmed by karyotyping.[2]
Treatment is based on symptoms.[2] Wilms' tumor is generally treated with surgery and chemotherapy, intellectual disabilities may be supported by special education, and kidney problems may be managed with ACE inhibitors or a kidney transplant.[2] It is believed that life expectancy is reduced, but it is not clear by how much.[3]
WAGR syndrome affects between 1 in 500,000 to 1 in a million people.[1] Males are affected more often than females.[2] The condition was first described by Miller in 1964.[4][5] The associated genetic abnormality was discovered in 1978 by Riccardi.[3]
Signs and symptoms
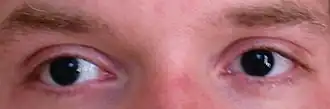
Newborn children with WAGR syndrome are soon noted to have aniridia. The clinical suspicion for WAGR may be increased with the presence of other genital anomalies, though genitourinary anomalies are not always present, particularly in girls.
Other common eye defects include cataracts and ptosis. About 50% of people develop Wilms' tumour.
The acronym WAGRO (O for obesity) has also been used and coinciding with loss of BDNF a gene that is also on chromosome 11.[6][7]
Cause
WAGR syndrome is caused by a mutation on chromosome 11 in the 11p13 region.[6] Specifically, several genes in this area are deleted, including the PAX6 ocular development gene and the Wilms' tumour gene (WT1).[8] Abnormalities in WT1 may also cause genitourinary anomalies. Mutations in the PAX6 gene have recently been shown to not only cause ocular abnormalities, but also problems in the brain and pancreas.[8][9][10][11]
The gene for brain-derived neurotrophic factor (BDNF), located on 11p14.1, has been proposed as a candidate gene for the obesity and excessive eating in a subset of WAGR patients.[12] This strengthens the case for a role for BDNF in energy balance.
Diagnosis
In older children, diagnosis of the syndrome can be made when aniridia and one of the other features are present. While aniridia is rarely absent in WAGR syndrome, cases have been reported without it. Chromosomal analysis is necessary for definitive diagnosis.[13][14]
Treatment
Children with WAGR syndrome receive regular (3-4 yearly) kidney surveillance for Wilms' tumour until at least the age of 6–8 years and thereafter remain under some follow-up because of the risk of late onset nephropathy (40% of patients with WAGR Syndrome over the age of 12 years). Females with WAGR syndrome may have streak ovaries, which can increase the risk for gonadoblastoma. Malformations of the vagina and/or uterus may also be present.
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 "WAGR syndrome: MedlinePlus Genetics". medlineplus.gov. Genetics Home Reference. Archived from the original on 11 December 2020. Retrieved 25 January 2021.
- 1 2 3 4 5 6 7 8 "WAGR Syndrome/11p Deletion Syndrome". NORD (National Organization for Rare Disorders). Archived from the original on 24 October 2020. Retrieved 26 January 2021.
- 1 2 3 Cassidy, Suzanne B.; Allanson, Judith E. (2010). Management of Genetic Syndromes. John Wiley & Sons. p. 897. ISBN 978-0-470-89314-2. Archived from the original on 2021-08-27. Retrieved 2021-01-26.
- ↑ Miller RW, Fraumeni JF, Manning MD (1964). "Association of Wilms's tumour with aniridia, hemihypertrophy and other congenital malformations". N Engl J Med. 270 (18): 922–7. doi:10.1056/NEJM196404302701802. PMID 14114111.
- ↑ Fazzi, Elisa; Bianchi, Paolo Emilio (2016). Visual Impairments and Developmental Disorders: From diagnosis to rehabilitation. John Libbey Eurotext. p. PT252. ISBN 978-2-7420-1482-8. Archived from the original on 2021-08-27. Retrieved 2021-01-26.
- 1 2 Online Mendelian Inheritance in Man (OMIM): WAGR syndrome - 194072
- ↑ Han JC, Liu QR, Jones M, Levinn RL, Menzie CM, Jefferson-George KS, Adler-Wailes DC, Sanford EL, Lacbawan FL, Uhl GR, Rennert OM, Yanovski JA (August 2008). "Brain-derived neurotrophic factor and obesity in the WAGR syndrome". The New England Journal of Medicine. 359 (9): 918–27. doi:10.1056/NEJMoa0801119. PMC 2553704. PMID 18753648.
- 1 2 Glaser T, Jepeal L, Edwards J, Young S, Favor J, Maas R (1994). "PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects". Nat Genet. 7 (4): 463–71. doi:10.1038/ng0894-463. PMID 7951315.
- ↑ Yasuda T, Kajimoto Y, Fujitani Y, Watada H, Yamamoto S, Watarai T, Umayahara Y, Matsuhisa M, Gorogawa S, Kuwayama Y, Tano Y, Yamasaki Y, Hori M (2002). "PAX6 mutation as a genetic factor common to aniridia and glucose intolerance". Diabetes. 51 (1): 224–30. doi:10.2337/diabetes.51.1.224. PMID 11756345.
- ↑ Mitchell T, Free S, Williamson K, Stevens J, Churchill A, Hanson I, Shorvon S, Moore A, van Heyningen V, Sisodiya S (2003). "Polymicrogyria and absence of pineal gland due to PAX6 mutation". Ann Neurol. 53 (5): 658–63. doi:10.1002/ana.10576. PMID 12731001.
- ↑ Talamillo A, Quinn J, Collinson J, Caric D, Price D, West J, Hill R (2003). "Pax6 regulates regional development and neuronal migration in the cerebral cortex". Dev Biol. 255 (1): 151–63. doi:10.1016/S0012-1606(02)00046-5. PMID 12618140.
- ↑ Han JC, Liu QR, Jones M, et al. (August 2008). "Brain-derived neurotrophic factor and obesity in the WAGR syndrome". N. Engl. J. Med. 359 (9): 918–27. doi:10.1056/NEJMoa0801119. PMC 2553704. PMID 18753648.
- ↑ Fischbach BV, Trout KL, Lewis J, Luis CA, Sika M (2005). "WAGR syndrome: a clinical review of 54 cases". Pediatrics. 116 (4): 984–8. doi:10.1542/peds.2004-0467. PMID 16199712.
- ↑ Turleau C, de Grouchy J, Nihoul-Fékété C, Dufier J, Chavin-Colin F, Junien C (1984). "Del11p13/nephroblastoma without aniridia". Hum Genet. 67 (4): 455–6. doi:10.1007/BF00291410. PMID 6092262.
External links
| Classification |
|
|---|---|
| External resources |
- DECIPHER database entry for 11p deletion syndrome