Ribonucleotide reductase
| ribonucleoside-diphosphate reductase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
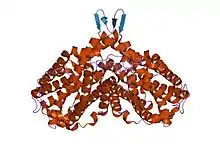
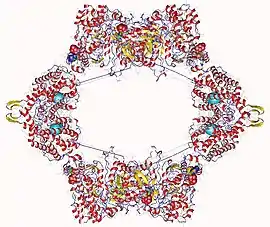 Ribonucleoside-diphosphate reductase heterooctamer, E.Coli | |||||||||
| Identifiers | |||||||||
| EC no. | 1.17.4.1 | ||||||||
| CAS no. | 9047-64-7sy | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
Ribonucleotide reductase (RNR), also known as ribonucleoside diphosphate reductase (rNDP), is an enzyme that catalyzes the formation of deoxyribonucleotides from ribonucleotides.[1] It catalyzes this formation by removing the 2'-hydroxyl group of the ribose ring of nucleoside diphosphates. This reduction produces deoxyribonucleotides.[2] Deoxyribonucleotides in turn are used in the synthesis of DNA. The reaction catalyzed by RNR is strictly conserved in all living organisms.[3] Furthermore, RNR plays a critical role in regulating the total rate of DNA synthesis so that DNA to cell mass is maintained at a constant ratio during cell division and DNA repair.[4] A somewhat unusual feature of the RNR enzyme is that it catalyzes a reaction that proceeds via a free radical mechanism of action.[5][6] The substrates for RNR are ADP, GDP, CDP and UDP. dTDP (deoxythymidine diphosphate) is synthesized by another enzyme (thymidylate kinase) from dTMP (deoxythymidine monophosphate).
Structure
Ribonucleotide reductases are divided into three classes. Class I RNR enzymes are constructed from large alpha subunit and small beta subunits which associate to form an active heterodimeric tetramer. By reducing NDPs to 2'-dNDPs, the enzyme catalyses the de novo synthesis of deoxyribonucleotides (dNTPs), which are precursors to DNA synthesis and essential for cell proliferation.[7] Class II RNRs produce a 5'-deoxyadenosyl radical by homolytic cleavage of the C-Co bond in adenosylcobalamin. In addition, Class III RNRs contain a stable glycyl radical.[8]
Humans carry Class I RNRs. The alpha subunit is encoded by the RRM1 gene while there are two isoforms of the beta subunit, encoded by the RRM2 and RRM2B genes:
|
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Each Class I alpha monomer consists of three domains:[9]
- one mainly helical domain comprising the 220 N-terminal residues,
- a second large ten-stranded α/β structure comprising 480 residues,
- and a third small five-stranded α/β structure comprising 70 residues.
In Pfam, the second domain has been interpreted as two separate domains:
- a shorter all-alpha N-terminal domain,
- and a longer barrel C-terminal domain.
|
|
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The Class I beta subunit usually contains a di-metal center and a stable tyrosyl radical. In humans, the beta subunit relies on a di-iron cofactor. In E. coli, the tyrosyl radical is located at position 122 (Y122) providing the stable radical for the Class I RNR2 subunits.[13] In A. aegypti, this tyrosyl radical is located at position 184 (Y184).[14] The tyrosyl radical is deeply buried inside the protein in a hydrophobic environment, located close to the iron center that is used in the stabilization of a tyrosyl radical. The structure of two μ-oxo-linked irons is dominated by ligands that serve as iron binding sites: four carboxylates [aspartate (D146), glutamate (E177, E240, and E274)] and two histidines (H180 and H277).[14] Association occurs between the C-terminus of RNR2 and the C-terminus of RNR1.[9] Enzymatic activity is dependent on association of the RNR1 and RNR2 subunits. The active site consists of the active dithiol groups from the RNR1 as well as the diferric center and the tyrosyl radical from the RNR2 subunit.
Other residues of RNR2, such as aspartate (D273), tryptophan (W48), and tyrosine (Y356) further stabilize the active-site tyrosyl radical thus allowing electron transfer.[9] These residues help in the transfer of the radical electron from tyrosine (Y122) of RNR2 to cysteine (C439) of RNR1. The electron transfer begins on RNR2 tyrosine (Y122) and continues in RNR2 to tryptophan (W48), which is separated from RNR1 tyrosine (Y731) by 2.5 nanometers. Electron transfer from RNR2 to RNR1 occurs via tyrosine (Y356 to Y731) and continues on through tyrosine (Y730) to cysteine (C439) in the active site.[15] Site-directed mutations of the RNR primary structure indicate that all residues cited above participate in the long distance transfer of the free radical to the active site.[9]
In A. aegypti mosquitoes, RNR1 retains most of the crucial amino acid residues, including aspartate (D64) and valine (V292 or V284), that are necessary in allosteric regulation; proline (P210 and P610), leucine (L453 and L473), and methionine (M603) residues that are located in the hydrophobic active site; cysteine (C225, C436 and C451) residues that are involved in removal of a hydrogen atom and transfer of the radical electron at the active site; cysteine (C225 and C436), asparagine (N434), and glutamate (E441) residues that bind the ribonucleotide substrate; tyrosine (Y723 and Y743) residues that dictate the radical transfer; and cysteine (C838 and C841) residues that are used in the regeneration of dithiol groups in the active site.[14]
Function
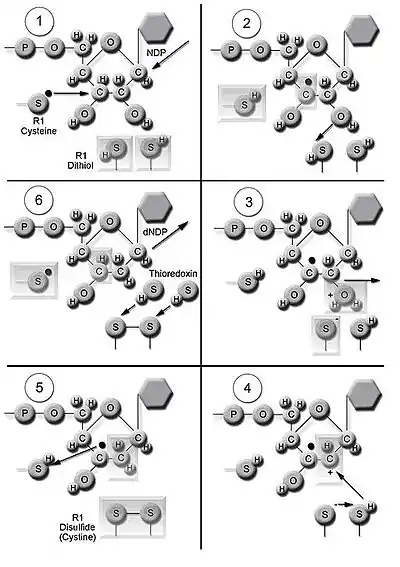
The enzyme ribonucleotide reductase (RNR) catalyzes the de novo synthesis of dNDPs.[16] Catalysis of ribonucleoside 5’-diphosphates (NDPs) involves a reduction at the 2’-carbon of ribose 5-phosphate to form the 2’-deoxy derivative-reduced 2’-deoxyribonucleoside 5’-diphosphates (dNDPs). This reduction is initiated with the generation of a free radical. Following a single reduction, RNR requires electrons donated from the dithiol groups of the protein thioredoxin. Regeneration of thioredoxin occurs when nicotinamide adenine dinucleotide phosphate (NADPH) provides two hydrogen atoms that are used to reduce the disulfide groups of thioredoxin.
Three classes of RNR have similar mechanisms for the reduction of NDPs, but differ in the domain that generates the free radical, the specific metal in the metalloprotein structure, and the electron donors. All classes use free-radical chemistry.[9] Class I reductases use an iron center with ferrous to ferric conversion to generate a tyrosyl free radical. Reduction of NDP substrates occurs under aerobic conditions. Class I reductases are divided into IA and IB due to differences in regulation. Class IA reductases are distributed in eukaryotes, eubacteria, bacteriophages, and viruses. Class IB reductases are found in eubacteria. Class IB reductases can also use a radical generated with the stabilization of a binuclear manganese center. Class II reductases generate the free radical 5’-deoxyadenosyl radical from cobalamin (coenzyme B12) and have a simpler structure than class I and class III reductases. Reduction of NDPs or ribonucleotide 5’-triphosphates (NTPs) occurs under either aerobic or anaerobic conditions. Class II reductases are distributed in archaebacteria, eubacteria, and bacteriophages. Class III reductases use a glycine radical generated with the help of an S-adenosyl methionine and an iron sulphur center. Reduction of NTPs is limited to anaerobic conditions. Class III reductases are distributed in archaebacteria, eubacteria, and bacteriophages.[9][14] Organisms are not limited to having one class of enzymes. For example, E. coli have both class I and class III RNR.
Catalytic reduction mechanism
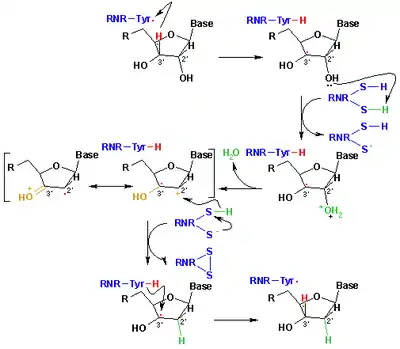
The mechanism that is currently accepted for the reduction of ribonucleotides to deoxyribonucleotides is depicted in the following scheme. The first step involves the abstraction of the 3’- H of substrate 1 by radical Cys439. Subsequently, the reaction involves the elimination of one water molecule from carbon C-2’ of the ribonucleotide, catalyzed by Cys225 and Glu441. In the third step there is a hydrogen atom transfer from Cys225 to carbon C-2’ of the 2’-ketyl radical 3, after previous proton transfer from Cys462 to Cys225. At the end of this step, a radical anionic disulfide bridge and the closed-shell ketone intermediate 4 are obtained. This intermediate has been identified during the conversion of several 2’-substituted substrate analogues, as well as with the natural substrate[17] interacting with enzyme mutants. The next step is the oxidation of the anionic disulfide bridge, with concomitant reduction of the substrate, generating 5. The spin density shifts from the sulphur atoms to the C-3' atom of the substrate, with simultaneous proton transfer from Glu441 to carbon C-3'. The last step is the reverse of the first step and involves a hydrogen transfer from Cys439 to C-3’, regenerating the initial radical and resulting in the final product 6.
Theoretical models of some steps of these mechanisms using the full model of the R1 protein can be found at the studies performed by Cerqueira et al..[18][19]
Regulation

Class I RNR comprises RNR1 and RNR2 subunits, which can associate to form a heterodimeric tetramer.[5] RNR1 contains both allosteric sites, mediating regulation of substrate specificity and activity.[11] Depending on the allosteric configuration, one of the four ribonucleotides binds to the active site.
Regulation of RNR is designed to maintain balanced quantities of dNTPs. Binding of effector molecules either increases or decreases RNR activity. When ATP binds to the allosteric activity site, it activates RNR. In contrast, when dATP binds to this site, it deactivates RNR.[9] In addition to controlling activity, the allosteric mechanism also regulates the substrate specificity and ensures the enzyme produces an equal amount of each dNTP for DNA synthesis.[9] In all classes, binding of ATP or dATP to the allosteric site induces reduction of cytidine 5’-diphosphate (CDP) and uridine 5’-diphosphate (UDP); 2’-deoxyguanosine 5’-triphosphate (dGTP) induces reduction of adenosine 5’-diphosphate (ADP); and 2’-deoxythymidine 5’-triphosphate (dTTP) induces reduction of guanosine 5’-diphosphate (GDP) (Figure 1).
Class IB reductases are not inhibited by dATP because they lack approximately 50 N-terminal amino acids required for the allosteric activity site.[20] Additionally, it is important that the activity of ribonucleotide reductase be under transcriptional and post-transcriptional control because the synthesis of damage-free DNA relies on a balanced pool of deoxyribonucleotides.[21] Eukaryotic cells with class IA reductases have a mechanism of negative control to turn off synthesis of dNTPs as they accumulate. This mechanism protects the cell from toxic and mutagenic effects that can arise from the overproduction of dNTPs because changes in balanced dNTP pools lead to DNA damage and cell death.[22][23] Although, the overproduction of dNTPs or an unbalanced supply of them can lead to misincorporation of nucleotides into DNA, the supply of dNTPs supply can allow for DNA repair. p53R2 is a small subunit of ribonucleotide reductase that can induce such repair. Changes within this p53 induced R2 homolog can cause depletion in mitochondrial DNA and consequently p53R2 serves a major factor in dNTP supply.[24]
RNR may use the morpheein model of allosteric regulation.[25]
RNR1 and RNR2 inhibitors
Generally Class I RNR inhibitors can be divided in three main groups: translation inhibitors, which block the synthesis of the enzyme; dimerization inhibitors that prevent the association of the two RNR subunits (R1 and R2); and catalytic inhibitors that inactivate the subunit R1 and/or subunit R2.[18]
Class I RNR can be inhibited by peptides similar to the C-terminus of RNR2. These peptides can compete with RNR2 for binding to RNR1, and as a result RNR1 does not form an enzymatically active complex with RNR2.[26][27] Although the C-terminus of RNR2 proteins is different across species, RNR2 can interact with RNR1 across species.[28] When the mouse RNR2 C-terminus was replaced with the E. coli RNR2 C-terminal (7 or 33) amino acid residues, the chimeric RNR2 subunit still binds to mouse RNR1 subunits. However, they lack enzymatic activity due probably to the elimination of residues involved in the transfer of the free radical electron from the RNR2 to the RNR1 subunit.[27]
Small peptides can specifically inhibit the RNR2 subunits from binding with RNR1 when they share a significant similarity with the normal RNR2 C-terminus.[29] This inhibition RNR2 binding to RNR1 has been tested successfully in herpes simplex virus (HSV) RNR. When a 7 amino acid oligomer (GAVVNDL) truncated from the C-terminus of the RNR2 subunit was used in competition assays, it prevented the normal RNR2 from forming an enzymatically active complex with RNR1.[30] Other small peptide inhibitors similar to the RNR2 C-terminus have also been used successfully to inhibit HSV RNR enzymatic activity and thus HSV replication.[31] In mice models of stromal keratitis and corneal neovascularization (HSV ocular disease), a small RNR2 C-terminal analog BILD 1263 has been reported to inhibit RNR and is effective in preventing these diseases.[32] In some cases, although treatment with small C-terminal analogs may not stop disease spreading, they can still help in healing. In the acyclovir-resistant HSV (PAAr5), a small peptide inhibitor BILD 1633 has been reported to be 5 to 10 times more potent than BILD 1263 against cutaneous PAAr5 infection.[33] A combination therapy approach (BILD 1633 and acyclovir) is more effective to heal topical lesions in mice. These data suggest that small peptide inhibitors that compete with RNR2 for binding to RNR1 are useful in preventing the spread of HSV.
Gallium inhibits RNR2 by substituting for Fe3+ in the active site. Gallium maltolate is an orally bioavailable form of gallium that exploits this inhibitory activity to treat cancer, infections, and other diseases.[34]
The drugs hydroxyurea[35] and Motexafin gadolinium interfere with the action of this enzyme.[36]
References
- ↑ Elledge SJ, Zhou Z, Allen JB (March 1992). "Ribonucleotide reductase: regulation, regulation, regulation". Trends in Biochemical Sciences. 17 (3): 119–23. doi:10.1016/0968-0004(92)90249-9. PMID 1412696.
- ↑ Sneeden JL, Loeb LA (September 2004). "Mutations in the R2 subunit of ribonucleotide reductase that confer resistance to hydroxyurea". The Journal of Biological Chemistry. 279 (39): 40723–8. doi:10.1074/jbc.M402699200. PMID 15262976.
- ↑ Torrents E, Aloy P, Gibert I, Rodríguez-Trelles F (August 2002). "Ribonucleotide reductases: divergent evolution of an ancient enzyme". Journal of Molecular Evolution. 55 (2): 138–52. doi:10.1007/s00239-002-2311-7. PMID 12107591. S2CID 24603578.
- ↑ Herrick J, Sclavi B (January 2007). "Ribonucleotide reductase and the regulation of DNA replication: an old story and an ancient heritage". Molecular Microbiology. 63 (1): 22–34. doi:10.1111/j.1365-2958.2006.05493.x. PMID 17229208. S2CID 9473163.
- 1 2 Eklund H, Eriksson M, Uhlin U, Nordlund P, Logan D (August 1997). "Ribonucleotide reductase—structural studies of a radical enzyme". Biological Chemistry. 378 (8): 821–5. doi:10.1515/bchm.1997.378.8.815. PMID 9377477.
- ↑ Stubbe J, Riggs-Gelasco P (November 1998). "Harnessing free radicals: formation and function of the tyrosyl radical in ribonucleotide reductase". Trends in Biochemical Sciences. 23 (11): 438–43. doi:10.1016/S0968-0004(98)01296-1. PMID 9852763.
- ↑ Fairman JW, Wijerathna SR, Ahmad MF, Xu H, Nakano R, Jha S, Prendergast J, Welin RM, Flodin S, Roos A, Nordlund P, Li Z, Walz T, Dealwis CG (March 2011). "Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization". Nature Structural & Molecular Biology. 18 (3): 316–22. doi:10.1038/nsmb.2007. PMC 3101628. PMID 21336276.
- ↑ Larsson KM, Jordan A, Eliasson R, Reichard P, Logan DT, Nordlund P (November 2004). "Structural mechanism of allosteric substrate specificity regulation in a ribonucleotide reductase". Nature Structural & Molecular Biology. 11 (11): 1142–9. doi:10.1038/nsmb838. PMID 15475969. S2CID 1025702.
- 1 2 3 4 5 6 7 8 Jordan A, Reichard P (1998). "Ribonucleotide reductases". Annual Review of Biochemistry. 67 (1): 71–98. doi:10.1146/annurev.biochem.67.1.71. PMID 9759483.
- 1 2 PDB: 1PEU; Uppsten M, Färnegårdh M, Jordan A, Eliasson R, Eklund H, Uhlin U (June 2003). "Structure of the large subunit of class Ib ribonucleotide reductase from Salmonella typhimurium and its complexes with allosteric effectors". Journal of Molecular Biology. 330 (1): 87–97. doi:10.1016/S0022-2836(03)00538-2. PMID 12818204.
- 1 2 Uhlin U, Eklund H (August 1994). "Structure of ribonucleotide reductase protein R1". Nature. 370 (6490): 533–9. doi:10.1038/370533a0. PMID 8052308. S2CID 8940689.
- ↑ Nordlund P, Eklund H (July 1993). "Structure and function of the Escherichia coli ribonucleotide reductase protein R2". Journal of Molecular Biology. 232 (1): 123–64. doi:10.1006/jmbi.1993.1374. PMID 8331655.
- ↑ Högbom M, Andersson ME, Nordlund P (March 2001). "Crystal structures of oxidized dinuclear manganese centres in Mn-substituted class I ribonucleotide reductase from Escherichia coli: carboxylate shifts with implications for O2 activation and radical generation". Journal of Biological Inorganic Chemistry. 6 (3): 315–23. doi:10.1007/s007750000205. PMID 11315567. S2CID 20748553.
- 1 2 3 4 Pham DQ, Blachuta BJ, Nichol H, Winzerling JJ (September 2002). "Ribonucleotide reductase subunits from the yellow fever mosquito, Aedes aegypti: cloning and expression". Insect Biochemistry and Molecular Biology. 32 (9): 1037–44. doi:10.1016/S0965-1748(02)00041-3. PMID 12213240.
- ↑ Chang MC, Yee CS, Stubbe J, Nocera DG (May 2004). "Turning on ribonucleotide reductase by light-initiated amino acid radical generation". Proceedings of the National Academy of Sciences of the United States of America. 101 (18): 6882–7. doi:10.1073/pnas.0401718101. PMC 406436. PMID 15123822.
- ↑ Cox M, Nelson DR (2008). Lehninger Principles of Biochemistry. San Francisco: W. H. Freeman. ISBN 978-0-7167-7108-1.
- ↑ Cerqueira NM, Fernandes PA, Eriksson LA, Ramos MJ (December 2004). "Ribonucleotide activation by enzyme ribonucleotide reductase: understanding the role of the enzyme". Journal of Computational Chemistry. 25 (16): 2031–7. doi:10.1002/jcc.20127. PMID 15481089. S2CID 19665974.
- 1 2 Cerqueira NM, Pereira S, Fernandes PA, Ramos MJ (2005). "Overview of ribonucleotide reductase inhibitors: an appealing target in anti-tumour therapy". Current Medicinal Chemistry. 12 (11): 1283–94. doi:10.2174/0929867054020981. PMID 15974997.
- ↑ Cerqueira NM, Fernandes PA, Eriksson LA, Ramos MJ (March 2006). "Dehydration of ribonucleotides catalyzed by ribonucleotide reductase: the role of the enzyme". Biophysical Journal. 90 (6): 2109–19. doi:10.1529/biophysj.104.054627. PMC 1386789. PMID 16361339.
- ↑ Eliasson R, Pontis E, Jordan A, Reichard P (October 1996). "Allosteric regulation of the third ribonucleotide reductase (NrdEF enzyme) from enterobacteriaceae". The Journal of Biological Chemistry. 271 (43): 26582–7. doi:10.1074/jbc.271.43.26582. PMID 8900130.
- ↑ Thelander L (June 2007). "Ribonucleotide reductase and mitochondrial DNA synthesis". Nature Genetics. 39 (6): 703–4. doi:10.1038/ng0607-703. PMID 17534360. S2CID 22565931.
- ↑ Kunz BA (1988). "Mutagenesis and deoxyribonucleotide pool imbalance". Mutation Research. 200 (1–2): 133–47. doi:10.1016/0027-5107(88)90076-0. PMID 3292903.
- ↑ Meuth M (April 1989). "The molecular basis of mutations induced by deoxyribonucleoside triphosphate pool imbalances in mammalian cells". Experimental Cell Research. 181 (2): 305–16. doi:10.1016/0014-4827(89)90090-6. PMID 2647496.
- ↑ Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, Chrétien D, de Lonlay P, Paquis-Flucklinger V, Arakawa H, Nakamura Y, Munnich A, Rötig A (June 2007). "Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion". Nature Genetics. 39 (6): 776–80. doi:10.1038/ng2040. PMID 17486094. S2CID 22103978.
- ↑ Selwood T, Jaffe EK (March 2012). "Dynamic dissociating homo-oligomers and the control of protein function". Archives of Biochemistry and Biophysics. 519 (2): 131–43. doi:10.1016/j.abb.2011.11.020. PMC 3298769. PMID 22182754.
- ↑ Climent I, Sjöberg BM, Huang CY (May 1991). "Carboxyl-terminal peptides as probes for Escherichia coli ribonucleotide reductase subunit interaction: kinetic analysis of inhibition studies". Biochemistry. 30 (21): 5164–71. doi:10.1021/bi00235a008. PMID 2036382.
- 1 2 Hamann CS, Lentainge S, Li LS, Salem JS, Yang FD, Cooperman BS (March 1998). "Chimeric small subunit inhibitors of mammalian ribonucleotide reductase: a dual function for the R2 C-terminus?". Protein Engineering. 11 (3): 219–24. doi:10.1093/protein/11.3.219. PMID 9613846.
- ↑ Cosentino G, Lavallée P, Rakhit S, Plante R, Gaudette Y, Lawetz C, Whitehead PW, Duceppe JS, Lépine-Frenette C, Dansereau N (January 1991). "Specific inhibition of ribonucleotide reductases by peptides corresponding to the C-terminal of their second subunit". Biochemistry and Cell Biology. 69 (1): 79–83. doi:10.1139/o91-011. PMID 2043345.
- ↑ Cooperman BS (2003). "Oligopeptide inhibition of class I ribonucleotide reductases". Biopolymers. 71 (2): 117–31. doi:10.1002/bip.10397. PMID 12767114. S2CID 25196379.
- ↑ Filatov D, Ingemarson R, Gräslund A, Thelander L (August 1992). "The role of herpes simplex virus ribonucleotide reductase small subunit carboxyl terminus in subunit interaction and formation of iron-tyrosyl center structure". The Journal of Biological Chemistry. 267 (22): 15816–22. doi:10.1016/S0021-9258(19)49608-7. PMID 1322407.
- ↑ Cohen EA, Gaudreau P, Brazeau P, Langelier Y (1986). "Specific inhibition of herpesvirus ribonucleotide reductase by a nonapeptide derived from the carboxy terminus of subunit 2". Nature. 321 (6068): 441–3. doi:10.1038/321441a0. PMID 3012360. S2CID 4238076.
- ↑ Brandt CR, Spencer B, Imesch P, Garneau M, Déziel R (May 1996). "Evaluation of a peptidomimetic ribonucleotide reductase inhibitor with a murine model of herpes simplex virus type 1 ocular disease". Antimicrobial Agents and Chemotherapy. 40 (5): 1078–84. doi:10.1128/aac.40.5.1078. PMC 163269. PMID 8723444.
- ↑ Duan J, Liuzzi M, Paris W, Lambert M, Lawetz C, Moss N, Jaramillo J, Gauthier J, Déziel R, Cordingley MG (July 1998). "Antiviral activity of a selective ribonucleotide reductase inhibitor against acyclovir-resistant herpes simplex virus type 1 in vivo". Antimicrobial Agents and Chemotherapy. 42 (7): 1629–35. doi:10.1128/aac.42.7.1629. PMC 105657. PMID 9660995.
- ↑ Bernstein LR (December 1998). "Mechanisms of therapeutic activity for gallium" (PDF). Pharmacological Reviews. 50 (4): 665–82. PMID 9860806.
- ↑ "Information on EC 1.17.4.1 – ribonucleoside-diphosphate reductase". Brenda. Retrieved 25 July 2015.
- ↑ Hashemy SI, Ungerstedt JS, Zahedi Avval F, Holmgren A (April 2006). "Motexafin gadolinium, a tumor-selective drug targeting thioredoxin reductase and ribonucleotide reductase". The Journal of Biological Chemistry. 281 (16): 10691–7. doi:10.1074/jbc.M511373200. PMID 16481328.
External links
- Ribonucleotide+reductases at the US National Library of Medicine Medical Subject Headings (MeSH)
- The ribonucleotide reductase database (RNRdb)