داء سيستيني
الداء السيستيني (بالإنجليزية: Cystinosis) هو أحد أمراض الاختزان في الجسميات الحالة التي تتميز بالتراكم غير الطبيعي للحمض الأميني السيستين.[1] وهو اضطراب وراثي يتبع نمطًا وراثيًا جسميًا متنحيًا. إنه اضطراب وراثي جسمي متنح نادر ينتج عن تراكم السيستين الحر في الجسيمات الحالة، مما يؤدي في النهاية إلى تكوين بلورات داخل الخلايا في جميع أنحاء الجسم. داء السيستين هو السبب الأكثر شيوعًا لمتلازمة فانكوني عند الأطفال. تحدث متلازمة فانكوني عندما تتعطل وظيفة الخلايا في الأنابيب الكلوية، مما يؤدي إلى إفراز كميات غير طبيعية من السكريات والأحماض الأمينية في البول، والتبول المفرط، وانخفاض مستويات البوتاسيوم والفوسفات في الدم.
| الداء السيستيني Cystinosis | |
|---|---|
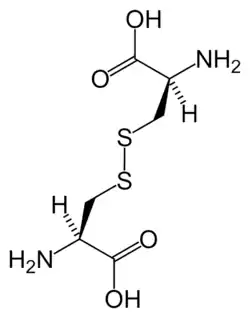 التركيب الكيميائي للسيستين في شكل ل-سيستين (تحت ظروف حيوية) التركيب الكيميائي للسيستين في شكل ل-سيستين (تحت ظروف حيوية) | |
| معلومات عامة | |
| الاختصاص | علم الغدد الصم |
| من أنواع | مرض اختزان في الجسميات الحالة، واضطراب جيني، واضطراب صبغي جسدي متنحي |
| الإدارة | |
| أدوية | سيستيامين ، وسيستيامين
|
كان داء السيستين أول مرض وراثي موثق ينتمي إلى مجموعة اضطرابات أمراض التخزين الليزوزومي.[2] يحدث داء السيستين بسبب طفرات في جين CTNS الذي يرمز السيستينوسين، الناقل الخاص بالغشاء الليزوزومي للسيستين. يتطلب استقلاب السيستين داخل الخلايا، مثلما يحدث مع جميع الأحماض الأمينية، نقله عبر غشاء الخلية. بعد تحلل البروتين الداخلي إلى السيستين داخل الجسيمات الحالة، ينتقل إلى العصارة الخلوية. ولكن إذا كان هناك خلل في البروتين الناقل، يتراكم السيستين في الجسيمات الحالة. نظرًا لأن السيستين غير قابل للذوبان، فعند زيادة تركيزه في الجسيمات الحالة داخل الأنسجة، تتشكل رواسب بلورية في جميع الأعضاء والأنسجة تقريبًا.[3]
ومع ذلك، لا يرتبط تطور المرض بوجود بلورات في الأنسجة المستهدفة. على الرغم من أن تلف الأنسجة قد ينتج عن ترسب السيستين، إلا أن آليات تلف الأنسجة ليست مفهومة تمامًا. تؤدي زيادة السيستين داخل الخلايا إلى اضطراب عملية التمثيل الغذائي للأكسدة الخلوية وحالة الغلوتاثيون،[4] مما يؤدي إلى تغيير استقلاب طاقة الميتوكندريون، والالتهام الذاتي، والاستماتة.[5]
يُعالج عادة داء السيستين باستخدام السيستامين، والذي يوصف لتقليل تراكم السيستين داخل الجسم.[6] ومع ذلك، فإن اكتشاف آليات مُمْرِضة جديدة وتطوير نموذج حيواني للمرض قد يفتحان الاحتمالات لتطوير طرق علاج جديدة لتحسين الإنذار على المدى الطويل.[2]
الأعراض
يوجد ثلاثة أنواع مميزة من داء السيستين لكل منها أعراض مختلفة قليلاً: داء السيستين الكلوي، وداء السيستين الوسيط، وداء السيستين غير الكلوي أو العيني. يُظهر الرضع المصابون بالداء السيستيني الكلوي فشل نمو ومشاكل خاصة في الكلى (تسمى أحيانًا متلازمة فانكوني الكلوية). تؤدي مشاكل الكلى إلى فقدان معادن مهمة وأملاح وسوائل ومواد مغذية أخرى. لا يؤدي فقدان العناصر الغذائية إلى إعاقة النمو فحسب، بل قد يؤدي إلى عظام ضعيفة ومنحنية (كساح نقص الفوسفات)، خاصة في الساقين. تؤدي الاختلالات الغذائية في الجسم إلى زيادة التبول والعطش والجفاف وزيادة حموضة الدم (الحماض).
بحلول سن الثانية تقريبًا، قد تترسب بلورات السيستين في القرنية أيضًا. يؤدي تراكم هذه البلورات في العين إلى زيادة الحساسية للضوء (رهاب الضوء). في حال عدم تطبيق العلاج في وقت مبكر من المرجح أن يعاني الأطفال المصابون بداء السيستين من فشل كلوي كامل في سن العاشرة تقريبًا. قد يتأخر حدوث ذلك عند تطبيق العلاج حتى وصول المرضى إلى سن المراهقة أو سن الـ 20. تشمل العلامات والأعراض الأخرى التي قد يحدث لدى المرضى: فقد عضلي، وعمى، وعدم القدرة على البلع، وضعف التعرق، ونقص تصبغ الشعر والجلد، ومرض السكري، ومشاكل الغدة الدرقية والجهاز العصبي.
تتشابه علامات وأعراض داء السيستين الوسيط مع داء السيستين الكلوي، ولكنها تحدث في سن متأخرة. تبدأ أعراض داء السيستين الوسيط عادةً بين الثانية عشرة والخامسة عشر عامًا. يُعتبر القصور الكلوي وترسب البلورات في القرنية السمتين الأوليتين الرئيسيتين لهذا الاضطراب. إذا تُرك داء السيستين الوسيط دون علاج، فسيحدث فشل كلوي كامل، ولكن عادةً لا يحدث ذلك حتى أواخر سن المراهقة حتى منتصف العشرينات.
لا يعاني الأشخاص المصابون بداء السيستين العيني أو غير الكلوي عادةً من ضعف النمو أو خلل في وظائف الكلى. العرض الوحيد هو رهاب الضوء بسبب ترسب بلورات السيستين في القرنية.
التشكل البلوري وتمييزه
بلورات السيستين سداسية الشكل وعديمة اللون. لا توجد في كثير من الأحيان في البول القلوي بسبب قابليتها العالية للذوبان. قد يكون من الصعب تمييز البلورات عديمة اللون عن بلورات حمض البول التي تكون سداسية أيضًا. في ظل الفحص تحت المجهر المستقطب، تكون البلورات ثنائية الانكسار مع تداخل مستقطب في اللون.[7]
مراجع
- A. Gahl, William؛ Jess G. Thoene؛ Jerry A. Schneider (2002)، "Cystinosis"، N Engl J Med، 347 (2): 111–121، doi:10.1056/NEJMra020552، PMID 12110740.
- Nesterova G, Gahl WA. Cystinosis: the evolution of a treatable disease. Pediatr Nephrol 2012;28:51–9.
- Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med 2002;347:111-121.
- Kumar A, Bachhawat AK. A futile cycle, formed between two ATP-dependent γ-glutamyl cycle enzymes, γ-glutamyl cysteine synthetase and 5-oxoprolinase: the cause of cellular ATP depletion in nephrotic cystinosis?; J Biosci 2010;35:21–25.
- Park MA, Thoene JG. Potential role of apoptosis in development of the cystinotic phenotype. Pediatr Nephrol 2005;20:441–446.
- Besouw M, Masereeuw R, Van den Heuvel L et al. Cysteamine: an old drug with new potential. Drug Discov Today 2013.
- Spencer, Daniel، "Cystine"، CRYSTALS، Urinalysis (Texas Collaborative for Teaching Excellence)، مؤرشف من الأصل في 6 نوفمبر 2016، اطلع عليه بتاريخ 4 مارس 2012.
 بوابة طب
بوابة طب