تتابع الحمض النووي الريبي RNA
يستخدم RNA-Seq تسلسل الجيل التالي (NGS) للكشف عن وجود وكمية من الحمض النووي الريبي في عينة بيولوجية في لحظة معينة، وتحليل المتغير الخلوي المستمر. [2] [3]

على وجه التحديد، يسهل RNA-Seq القدرة على النظر إلى الجينات البديلة المقسمة، والتعديلات بعد النسخ، والانصهار الجيني، والطفرات / SNPs والتغيرات في التعبير الجيني مع مرور الوقت، أو الاختلافات في التعبير الجيني في مجموعات أو علاجات مختلفة. [4] بالإضافة إلى نسخ mRNA، يمكن أن ينظر RNA-Seq إلى مجموعات مختلفة من الحمض النووي الريبي (RNA) لتشمل إجمالي الحمض النووي الريبي (RNA)، والحمض النووي الريبي (RNA) الصغير، مثل miRNA، الحمض الريبي النووي النقال (tRNA)، وتحديد ملامح الريبوسوم. [5] يمكن أيضًا استخدام RNA-Seq لتحديد حدود exon / intron والتحقق من أو تعديل حدود الجين الموضحة مسبقًا 5 و3. تشمل التطورات الحديثة في RNA-Seq تسلسل خلية وحيدة وتسلسل الأنسجة الثابتة في الموقع. [6]
قبل RNA-Seq، أجريت دراسات التعبير الجيني مع المصفوفات الدقيقة القائمة على التهجين. تشمل مشاكل المصفوفات الدقيقة القطع الجينية المتقاطعة، التهميش السيئ للجينات منخفضة الوزن وعالية التعبير، والحاجة إلى معرفة التسلسل المسبق. [7] بسبب هذه المشكلات الفنية، انتقلت النصوص إلى الأساليب القائمة على التسلسل. لقد تطورت هذه من تسلسل Sanger في مكتبات Expressed Sequence Tag، إلى الأساليب القائمة على العلامات الكيميائية (على سبيل المثال، التحليل التسلسلي للتعبير الجيني)، وأخيراً إلى التكنولوجيا الحالية، تسلسل الجيل القادم من cDNA (خاصة RNA-Seq).
الأساليب
إعداد متسلسل
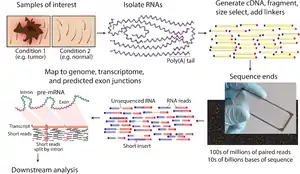
الخطوات العامة لإعداد متسلسل DNA (cDNA) التكميلية للتسلسل موضحة أسفل، ولكن غالبًا ما تختلف بين المنصات. [8] [3] [9]
- عزل الحمض النووي الريبي (RNA): يتم عزل الحمض النووي الريبي (RNA) من الأنسجة ويخلط مع (DNase) DNase يقلل من كمية الحمض النووي الجيني. يتم ضبط الكمية المفككة من الحمض النووي الريبي مع هلام والخلاصة التحليل الكهربي لمحتوى الشعيرات الدموية ويستخدم لتعيين عدد سلامة الحمض النووي الريبي للعينة. تؤخذ جودة الحمض النووي الريبي (RNA) والمبلغ الإجمالي لبدء الحمض النووي الريبي (RNA) في الاعتبار أثناء خطوات إعداد وتسلسل وتحليل المكتبة اللاحقة.
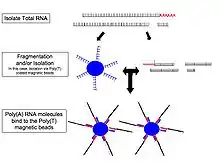
- اختيار / نضوب الحمض النووي الريبي: لتحليل إشارات الاهتمام، يمكن إما الاحتفاظ بالحمض النووي الريبي المعزول كما هو، مع ترشيح الحمض النووي الريبي مع ذيول متعددة الأقطاب (3) متعددة (poly (A) لتشمل فقط rRNA، المستخلص من الحمض النووي (rRNA)، و / أو تمت تصفيته من أجل الحمض النووي الريبي (RNA) الذي يربط تسلسلات محددة (جدول اختيار ونضج الحمض النووي الريبي، أسفل). الحمض النووي الريبي مع ذيول 3 '(بولي) الناضجة، والمعالجة، والتابعة لتسلسل الترميز. يتم اختيار (Poly A) عن طريق مزج الحمض النووي الريبي (RNA) مع قلة قليلة من القسيمات (T) المرتبطة تساهميًا بالركيزة، الخرز المغناطيسي عادة. [10] [11] يتجاهل اختيار (Poly A) الحمض النووي الريبي غير المشفر ويقدم تحيزًا 3 '، [12] والذي يتم تجنبه باستراتيجية نضوب الريبوسوم. تتم إزالة rRNA لأنه يمثل أكثر من 90 ٪ من الحمض النووي الريبي في الخلية، والتي إذا تم الاحتفاظ بها سوف تتخلص من البيانات الأخرى.
- تخليق cDNA: يتم نسخ RNA إلى cDNA لأن الحمض النووي أكثر استقرارًا ويسمح بالتضخيم (عن طريق polymerases الحمض النووي) وتقنية تسلسل الحمض النووي أكثر نضجًا. يؤدي التضخيم اللاحق للنسخ العكسي إلى فقد السبل، والتي يمكن تجنبها بالوسم الكيميائي أو تسلسل جزيء واحد. يتم تنفيذ تجزئة واختيار الحجم لتنقية التسلسلات التي هي الطول المناسب لآلة التسلسل. الحمض النووي الريبيrRNA، cDNA,أو كليهما مجزأ مع الإنزيمات، صوتنه، أو البخاخات. يعمل تجزئة الحمض النووي الريبي على تقليل انحياز 5 'من النسخ العكسي العشوائي وتأثير مواقع الربط التمهيدي، مع الجانب الذي ينتهي به الطرفان 5 و3 إلى الحمض النووي أقل كفاءة. يتبع التجزؤ اختيار الحجم، حيث تتم إزالة تسلسلات صغيرة أو تحديد نطاق ضيق من أطوال التسلسل. بسبب ضياع rRNA الصغيرة مثل miRNAs، يتم تحليلها بشكل مستقل. يمكن فهرسة cDNA لكل تجربة مع الباركود hexamer أو octamer، بحيث يمكن تجميع هذه التجارب في مسار واحد لتسلسل متعدد الإرسال.
| إستراتيجية | نوع الحمض النووي rRNA | محتوى الحمض النووي rRNA | محتوى الحمض النووي rRNA غير المعالج | محتوى الحمض النووي الجيني | طريقة عزل المحتوى |
|---|---|---|---|---|---|
| مجموع الحمض النووي rRNA | الكل | عالي | عالي | عالي | لا شيء |
| اختيار PolyA | مرموز | منخفض | منخفض | منخفض | تهجين مع oligomers بولي (dT) |
| نضوب rRNA | مرموز، غير مرموز | منخفض | عالي | عالي | زالة ال oligomers المكملة ل rRNA |
| الحصول على rRNA | مستهدف | منخفض | متوسط | منخفض | التهجين مع اجزاء مكملة للتابعات المطلوبة |
RNA صغير / غير مرموز لتسلسل RNA
عند إجراء تسلسل RNA غير miRNA، يتم تعديل إعداد التسلسل. يتم تحديد الحمض النووي الريبي على أساس نطاق الحجم المطلوب. لأهداف الحمض النووي الريبي الصغيرة، مثل miRNA، يتم عزل الحمض النووي الريبي من خلال اختيار الحجم. يمكن إجراء ذلك باستخدام هلام استبعاد الحجم، من خلال حبات مغناطيسية لاختيار الحجم، أو باستخدام مجموعة تم تطويرها تجاريًا. بمجرد عزلها، تتم إضافة الروابط إلى النهاية 3 'و5' ثم تنقيتها. والخطوة الأخيرة هي الجيل cDNA من خلال النسخ العكسي.
تسلسل الحمض النووي الريبي rRNA المباشر
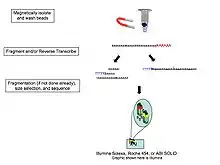
نظرًا لأن تحويل الحمض النووي الريبي إلى الحمض النووي الريبي النووي (cDNA)، فقد ثبت أن عمليات الربط، والتضخيم، والتلاعب بالعينات الأخرى تقدم تحيزات وأدوات أثرية قد تتداخل مع كل من التوصيف الصحيح والتابعات الصحيحة للنصوص الجينية، [13] تم استكشاف تسلسل الحمض النووي الريبي المباشر للجزيء المفرد [13] ، بما في ذلك Helicos (المفلسة) ، أوكسفورد نانوبور تكنولوجيز، [14] وغيرها. هذه التكنولوجيا تسلسل جزيئات الحمض النووي الريبي مباشرة بطريقة موازية على نطاق واسع.
تسلسل RNA أحادي الخلية (scRNA-Seq)
الطرق القياسية مثل المصفوفات الدقيقة وتحليل المعيار RNA-Seq يحلل تعبير ال rRNA من مجموعات كبيرة من الخلايا. في مجموعات الخلايا المختلطة، قد تحجب هذه القياسات الفروق الحرجة بين الخلايا الفردية داخل هذه المجموعات. [15] [16]
يوفر تسلسل الحمض النووي الريبي أحادي الخلية (scRNA-Seq) ملفات تعريف للخلايا الفردية. على الرغم من أنه لا يمكن الحصول على معلومات كاملة عن كل RNA الذي يعبر عن كل خلية، نظرًا للكمية الضئيلة من المواد المتاحة، يمكن التعرف على أنماط التعبير الجيني من خلال تحليلات تجميع الجينات. هذا يمكن أن يكشف عن وجود أنواع خلايا نادرة داخل مجتمع الخلية والتي ربما لم يسبق رؤيتها من قبل. على سبيل المثال، تم تحديد خلايا متخصصة نادرة في الرئة تدعى الخلايا الأيونية الرئوية التي تعبر عن منظم توصيل غشاء التليف الكيسي في 2018 من قبل مجموعتين تؤديان ال rRna ساق على ظهارة مجرى الرئة. [17] [18]
الإجراءات التجريبية
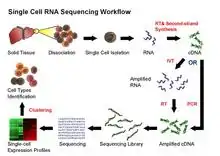
تتضمن بروتوكولات scRNA-Seq الحالية الخطوات التالية: عزل خلية واحدة وRNA، النسخ العكسي (RT)، والتضخيم، وتوليد التسلسل، والتسلسل. طرق مبكرة فصل الخلايا الفردية إلى آبار منفصلة. طرق أكثر حداثة تغلف الخلايا الفردية في قطرات في جهاز ميكروفلويديك، حيث يحدث رد فعل النسخ العكسي، وتحويل الحمض النووي الريبي إلى cDNAكل قطرة تحمل "الباركود" DNA الذي يسمي بشكل فريدcDNA المستمدة من خلية واحدة. بمجرد اكتمال النسخ العكسي، يمكن خلط cDNAمن العديد من الخلايا معًا للتسلسل؛ يتم التعرف على النصوص من خلية معينة بواسطة الرمز الشريطي الفريد. [19] [20]
تشمل تحديات scRNA-Seq الحفاظ على الوفرة النسبية الأولية للـ mRNA في الخلية وتحديد النصوص النادرة. [21] تعتبر خطوة النسخ العكسي أمرًا بالغ الأهمية نظرًا لأن كفاءة رد فعل RT تحدد مقدار تحليل الحمض النووي الريبي للخلية الذي سيتم تحليله في نهاية الأمر بواسطة جهاز التسلسل. قد تؤثر قابلية النسخ العكسي واستراتيجيات التحضير المستخدمة على إنتاج cDNA كامل الطول وتوليد مكتبات منحازة نحو نهاية 3 أو 5 'من الجينات.
في خطوة التضخيم، يتم الآن استخدام PCR أو في النسخ المختبرية (IVT) لتضخيم cDNA واحدة من مزايا الأساليب القائمة على PCR هي القدرة على توليد cDNA كامل الطول. ومع ذلك، قد يتم تضخيم كفاءة PCR مختلفة على تسلسلات معينة على سبيل المثال، محتوى GC وهيكل snapback بشكل كبير، مما ينتج مكتبات ذات تغطية غير متساوية. من ناحية أخرى، في حين أن التسلسلات التي تم إنشاؤها بواسطة IVT يمكنها تجنب تحيز التسلسل الناتج عن PCR، فقد يتم نسخ تسلسلات محددة بطريقة غير فعالة، مما يسبب تسلسل المتسلسل أو توليد تسلسلات غير مكتملة. [22] [15] تم نشر العديد من بروتوكولات scRNA-Seq: Tang et al.، STRT . ، [23] STRT ، [24] SMART-seq ، [25] CEL-seq ، [26] RAGE-seq ، [27] ، Quartz-seq. [28] و C1-CAGE. [29] من حيث استراتيجيات النسخ العكسي، وتوليف cDNA والتضخيم، وإمكانية استيعاب الباركود الخاصة بالتسلسل مثل UMIs أو القدرة على معالجة العينات المجمعة. [30]
في عام 2017، تم تقديم طريقتين لقياس في وقت واحد مرنا وحيد الخلية وتعبير البروتين من خلال الأجسام المضادة المسمى أليغنوكليوتيد المعروفة باسم REAP-seq ، [31] و CITE-seq. [32]
تطبيقات
أصبح scRNA-Seq يستخدم على نطاق واسع عبر التخصصات البيولوجية بما في ذلك التنمية، وعلم الأعصاب ، [33] علم الأورام ، [34] [35] [36] مرض المناعة الذاتية ، [37] والأمراض المعدية . [38]
قدمت scRNA-Seq نظرة ثاقبة كبيرة في تطور الأجنة والكائنات الحية، بما في ذلك دودة Caenorhabditis elegans ، [39] والمستوصف الشمسي التجديدي المتوسطي . [40] [41] كانت أول الحيوانات الفقارية التي تم تعيينها بهذه الطريقة هي الزرد [42] [43] و Xenopus laevis . [44] في كل حالة، تمت دراسة مراحل متعددة من الجنين، مما يسمح برسم عملية التنمية بأكملها على أساس كل خلية على حدة. [8] لقد أدرك العلم أن هذه التطورات هي " اختراق العام 2018 " . [45]
الاعتبارات التجريبية
يتم مراعاة مجموعة متنوعة من المعلمات عند تصميم وإجراء تجارب RNA-Seq:
- نوعية الأنسجة: يختلف التعبير الجيني داخل الأنسجة وفيما بينها، ويقيس RNA-Seq هذا المزيج من أنواع الخلايا. هذا قد يجعل من الصعب عزل الآلية البيولوجية للاهتمام. يمكن استخدام تسلسل خلية واحدة لدراسة كل خلية على حدة، وتخفيف هذه المشكلة.
- الاعتماد على الوقت: يتغير تعبير الجينات بمرور الوقت، ولا يأخذ RNA-Seq سوى لقطة. يمكن إجراء تجارب الدورة الزمنية لمراقبة التغييرات في النص الجيني.
- التغطية (المعروف أيضًا باسم العمق): يحتوي الحمض النووي الريبي على نفس الطفرات التي لوحظت في الحمض النووي، ويتطلب الكشف تغطية أعمق. مع تغطية عالية بما فيه الكفاية، يمكن استخدام RNA-Seq لتقدير التعبير عن كل أليل. قد يوفر هذا نظرة ثاقبة الظواهر مثل التأثيرات أو التأثيرات التنظيمية. يمكن استقراء عمق التسلسل المطلوب لتطبيقات محددة من تجربة تجريبية. [46]
- قطع لتوليد البيانات (تُعرف أيضًا باسم التباين التقني): يمكن أن تؤدي الكواشف (على سبيل المثال، مجموعة إعداد التسلسل) والموظفين المعنيين ونوع جهاز التسلسل على سبيل المثال، Illumina وPacific Biosciences إلى آثار فنية قد يساء تفسيرها على أنها نتائج ذات معنى. كما هو الحال مع أي تجربة علمية، من الحكمة إجراء RNA-Seq في بيئة جيدة التحكم. إذا لم يكن ذلك ممكنًا أو كانت الدراسة عبارة عن تحليل تلوي، فإن الحل الآخر هو اكتشاف القطع الفنية عن طريق استنتاج المتغيرات الكامنة (عادة تحليل المكون الرئيسي أو تحليل العوامل) والتصحيح لاحقًا لهذه المتغيرات. [47]
- إدارة البيانات: عادة ما تكون تجربة RNA-Seq في البشر في حدود 1 غيغابايت . [48] هذا الحجم الكبير من البيانات يمكن أن يطرح مشاكل التخزين. أحد الحلول هو ضغط البيانات باستخدام مخططات حسابية متعددة الأغراض (على سبيل المثال، gzip أو مخططات خاصة بالجينوم. هذا الأخير يمكن أن يكون على أساس تسلسل مرجعي أو دي نوفو. الحل الآخر هو إجراء تجارب ميكروأري، والتي قد تكون كافية لإجراء الدراسات التي تعتمد على الفرضيات أو تكرارها على عكس البحوث الاستكشافية.
التحليل

التجمع النسخي
يتم استخدام طريقتين لتعيين تسلسلات القراءة الأولية للميزات الجينومية (أي تجميع النص الجيني):
- دي نوفو: هذا النهج لا يحتاج إلى جينوم مرجعي لإعادة بناء النص، ويستخدم عادة إذا كان الجينوم غير معروف، غير مكتمل، أو تم تغييره بشكل كبير مقارنة بالمرجع. [49] تتضمن التحديات عند استخدام عمليات القراءة القصيرة لتجميع de novo 1)) تحديد القراءات التي يجب ضمها معًا في تسلسلات متجاورة contigs)، 2) متانة أخطاء التسلسل وغيرها من الأعمال الفنية، و3 الكفاءة الحسابية. تم نقل الخوارزمية الأساسية المستخدمة لتجميع de novo من الرسوم البيانية المتداخلة، والتي تحدد جميع التداخلات بين الزوجين إلى القراءات، إلى الرسوم البيانية لـ Bru Brunn، والتي تقسم القراءة إلى تسلسلات طولها k وتنهار كل k-mers في جدول تجزئة. [50] تم استخدام الرسوم البيانية المتداخلة مع تسلسل Sanger، لكن لا يتم القياس جيدًا إلى ملايين القراءات التي تم إنشاؤها باستخدام RNA-Seq. أمثلة على المجمعات التي تستخدم الرسوم البيانية دي Bruijn هي Velvet ، [51] Trinity ، Oases ، [52] و Bridger. [53] يمكن للنهاية المزدوجة وتسلسل القراءة الطويلة من نفس العينة أن يخففا من العجز في تسلسل القراءة القصيرة من خلال العمل كقالب أو هيكل عظمي. تشتمل مقاييس تقييم جودة مجموعة de novo على متوسط طول contig وعدد contigs و N50 . [54]

- دليل الجينوم: تعتمد هذه الطريقة على نفس الأساليب المستخدمة لمحاذاة الحمض النووي، مع التعقيد الإضافي المتمثل في محاذاة القراءات التي تغطي أجزاء غير مستمرة من الجينوم المرجعي. [55] هذه القراءات غير المستمرة هي نتيجة لتسلسل النصوص المقسمة (انظر الشكل). عادة، تحتوي خوارزميات المحاذاة على خطوتين: 1) محاذاة أجزاء قصيرة من القراءة (أي، بداية الجينوم) ، و 2) استخدام البرمجة الديناميكية لإيجاد محاذاة مثالية، وأحيانًا بالاقتران مع تعليقات توضيحية معروفة. تشمل أدوات البرمجيات التي تستخدم المحاذاة الموجهة للجينوم Bowtie ، [56] TopHat (الذي يبني على نتائج BowTie لمحاذاة الوصلات الفاصلة) ، [57] [58] Subread ، [59] STAR ، HISAT2 ، [60] Sailfish ، [61] كاليستو، [62] و GMAP. [63] يمكن قياس جودة التجميع الموجه للجينوم بمقاييس التجميع 1) de novo (على سبيل المثال، N50) و 2) مقارنات بالتسلسلات المعروفة، تقاطع الوصلات، الجينوم، والبروتين باستخدام الدقة ، الاستعادة ، أو توليفها (على سبيل المثال، النتيجة F1). [54] بالإضافة إلى ذلك، يمكن إجراء تقييم السيليكو باستخدام قراءات محاكية. [64] [65]
ملاحظة على جودة التجميع: الإجماع الحالي هو أن 1) يمكن أن تختلف جودة التجميع تبعًا للمقياس المستخدم، 2) التجميعات التي سجلت جيدًا في أحد الأنواع لا تؤدي بالضرورة أداءً جيدًا في الأنواع الأخرى، و3) قد يؤدي الجمع بين الطرق المختلفة إلى كن الأكثر موثوقية. [66] [67]
التعبير الجيني الكمي
يتم التعبير عن التعبير الكمي لدراسة التغيرات الخلوية استجابةً للمنبهات الخارجية، الاختلافات بين الحالات الصحية والمرضية، وغيرها من الأسئلة البحثية. غالبًا ما يستخدم تعبير الجين كبديل لوفرة البروتين، لكن غالبًا ما تكون هذه العناصر غير متكافئة بسبب أحداث ما بعد النسخ مثل تدخل الحمض النووي الريبي وانحلال بوساطة الهراء. [68]
يتم تحديد التعبير عن طريق حساب عدد القراءات التي تم تعيينها لكل موقع في خطوة التجميع transcriptome. يمكن قياس التعبير عن الإكسونات أو الجينات باستخدام contigs أو التعليقات التوضيحية المرجعية. [8] تم التحقق من صحة تعدادات RNA-Seq التي تمت ملاحظتها بقوة ضد التقنيات القديمة، بما في ذلك المصفوفات الدقيقة التعبير و qPCR . [46] [69] من الأمثلة على الأدوات التي تحدد التعدادات HTSeq ، [70] FeatureCounts ، [71] Rcount ، [72] maxcounts ، [73] FIXSEQ ، [74] و Cuffquant. ثم يتم تحويل عدد القراءة إلى مقاييس مناسبة لاختبار الفرضيات والانحدارات والتحليلات الأخرى. المعلمات لهذا التحويل هي:
- العمق / التغطية المتسلسلة: على الرغم من أن العمق محدد مسبقًا عند إجراء تجارب RNA-Seq متعددة، إلا أنه سيظل متباينًا على نطاق واسع بين التجارب. [75] لذلك، عادةً ما يتم تطبيع إجمالي عدد القراءات التي تم إنشاؤها في تجربة واحدة عن طريق تحويل التعدادات إلى أجزاء أو قراءات أو تعدادات لكل مليون قراءة معيّنة FPM أو RPM أو CPM يشار إلى عمق التسلسل أحيانًا بحجم المكتبة، وعدد جزيئات cDNA الوسيطة في التجربة.
- طول الجينات: سيكون للجينات الأطول شظايا / قراءات / تعدادات أكثر من الجينات الأقصر إذا كان تعبير النص الجيني هو نفسه. يتم ضبط هذا عن طريق تقسيم FPM على طول الجين، مما يؤدي إلى شظايا متري لكل كيلوبايت من النص لكل مليون قراءة المعينة (FPKM). [76] عند النظر إلى مجموعات من الجينات عبر العينات، يتم تحويل FPKM إلى نصوص لكل مليون (TPM) عن طريق تقسيم كل FPKM على مجموع FPKMs داخل عينة. [77] [78]
- مجموع عينة الحمض النووي الريبي: لأن نفس كمية الحمض النووي الريبي المستخرجة من كل عينة، فإن العينات ذات الحمض النووي الريبي الإجمالي أكثر سيكون لها عدد أقل من الحمض النووي الريبي لكل جين. يبدو أن هذه الجينات قد قللت من التعبير، مما أدى إلى إيجابيات كاذبة في التحليلات النهائية.
- تباين تعبير كل جين: يتم تصميمه ليتم حسابه على خطأ أخذ العينات (مهم للجينات ذات التعداد المنخفض للقراءة)، ويزيد الطاقة، ويقلل من الإيجابيات الخاطئة. يمكن تقدير التباين كتوزيع طبيعي، أو بواسون، أو سلبي ذو حدين [79] [80] [81] وغالبًا ما يتحلل إلى تباين تقني وبيولوجي.
الكم المطلق
الكم المطلق للتعبير الجيني غير ممكن في معظم تجارب RNA-Seq، والتي تقيس التعبير بالنسبة لجميع النصوص الجينية. من الممكن عن طريق إجراء RNA-Seq مع spike-ins، عينات من RNA بتركيزات معروفة. بعد التسلسل، يتم استخدام التعدادات المقروءة لتسلسلات الزيادة في تحديد العلاقة بين تعدادات القراءة لكل جين والكميات المطلقة من الأجزاء البيولوجية. [11] [82] في أحد الأمثلة، تم استخدام هذه التقنية في أجنة Xenopus المدارية لتحديد حركية النسخ. [83]
التعبير (النسخ) التفاضلي
إن الاستخدام البسيط والأقوى في كثير من الأحيان لـ RNA-Seq هو إيجاد اختلافات في التعبير الجيني بين شرطين أو أكثر (على سبيل المثال، المعالجة مقابل غير المعالجة)؛ وتسمى هذه العملية التعبير التفاضلي. يشار إلى المخرجات بشكل متكرر على أنها جينات معبر عنها تفاضليًا (DEGs) ويمكن أن يتم تنظيم هذه الجينات إما إلى أعلى أو أسفل (أي، أعلى أو أقل في حالة الاهتمام). هناك العديد من الأدوات التي تؤدي تعبيرًا تفاضليًا. يتم تشغيل معظمها في R أو Python أو سطر أوامر Unix . الأدوات الشائعة الاستخدام تشمل DESeq ، [80] edgeR ، [81] و voom + limma ، [79] [84] وكلها متوفرة من خلال موصل R / Bioconductor . [85] [86] هذه هي الاعتبارات الشائعة عند تنفيذ التعبير التفاضلي:
- المدخلات: تشمل مدخلات التعبير التفاضلي (1) مصفوفة تعبير RNA-Seq (جينات M X × عينات) و (2) مصفوفة تصميم تحتوي على ظروف تجريبية لعينات N. تحتوي أبسط مصفوفة التصميم على عمود واحد ، يقابل التسميات للحالة التي يجري اختبارها. يمكن أن تشمل المتغيرات الأخرى (يشار إليها أيضًا بعوامل أو ميزات أو تسميات أو معلمات) تأثيرات الدُفعات والمصنوعات المعروفة وأي بيانات وصفية قد تربك أو تتوسط في التعبير الجيني. بالإضافة إلى المتغيرات المشتركة المعروفة ، يمكن أيضًا تقدير المتغيرات المشتركة غير المعروفة من خلال أساليب التعلم الآلي غير الخاضعة للإشراف بما في ذلك تحليلات المكون الرئيسي والمتغير البديل [87] و PEER.[47] غالبًا ما تستخدم التحليلات المتغيرة المخفية لبيانات RNA-Seq الخاصة بالأنسجة البشرية ، والتي تحتوي عادةً على قطع أثرية إضافية لم يتم التقاطها في البيانات الوصفية (على سبيل المثال ، الوقت الإقفاري ، الاستعانة بمؤسسات متعددة ، السمات السريرية الأساسية ، جمع البيانات عبر العديد من السنوات مع العديد من الموظفين).
- الطرق: تستخدم معظم الأدوات إحصائيات الانحدار أو غير البارامترية لتحديد الجينات التي يتم التعبير عنها تفاضليًا ، وتكون إما قائمة على العد (DESeq2 ، limma ، edgeR) أو قائمة على التجميع (عن طريق القياس الكمي بدون محاذاة ، أو الكلب ، [88] [89] Cuffdiff ، [88] Ballgown [90] ). [91] بعد الانحدار ، تستخدم معظم الأدوات إما معدل الخطأ العائلي (FWER) أو تعديلات قيمة الاكتشاف الخاطئ (FDR) لمراعاة الفرضيات المتعددة (في الدراسات البشرية ، ~ 20000 من جينات ترميز البروتين أو ~ 50000 نموذج حيوي).
- المخرجات: يتكون المخرج النموذجي من صفوف تتوافق مع عدد الجينات وثلاثة أعمدة على الأقل، يتغير كل سجل من الجينات (تحويل سجل لنسبة التعبير في الظروف بين، مقياس حجم التأثير)، قيمة p، وp القيمة تعديلها لمقارنات متعددة. يتم تعريف الجينات على أنها ذات معنى من الناحية البيولوجية إذا نجحت في تحديد حجم التأثير (تغيير طية السجل) والأهمية الإحصائية. يجب تحديد هذه القطع بشكل مسبق بشكل مسبق، ولكن طبيعة تجارب RNA-Seq غالبًا ما تكون استكشافية، لذا يصعب التنبؤ بأحجام التأثير والقطع ذات الصلة في وقت مبكر.
- مطبات: سبب وجود هذه الأساليب المعقدة هو تجنب عدد لا يحصى من المزالق التي يمكن أن تؤدي إلى أخطاء إحصائية وتفسيرات مضللة. المطبات تشمل زيادة معدلات إيجابية كاذبة (بسبب المقارنات المتعددة)، والتحف إعداد العينات، وعدم تجانس العينة (مثل الخلفيات الوراثية المختلطة)، وعينات مترابطة للغاية، في عداد المفقودين للتصاميم التجريبية متعددة المستويات، والتصميم التجريبي الفقراء. أحد الأخطاء البارزة هو عرض النتائج في Microsoft Excel. [92] على الرغم من أنه مناسب، يقوم Excel تلقائيًا بتحويل بعض أسماء الجينات SEPT1 وDEC1 وMARCH2 إلى تواريخ أو أرقام الفاصلة العائمة.
- اختيار الأدوات والقياس: هناك العديد من الجهود التي تقارن نتائج هذه الأدوات، حيث تميل DESeq2 إلى التفوق بشكل معتدل على الطرق الأخرى. [93] [94] [95] [96] [97] [98] كما هو الحال مع الطرق الأخرى، يتكون القياس من مقارنة مخرجات الأداة مع بعضها البعض ومعاييرgolden المعروف.
تحاليل الدارجة للحصول على قائمة من الجينات المعبر عنها تفاضليًا تأتي في اثنين من النكهات، التحقق من صحة الملاحظات والاستدلال البيولوجي. نظرًا لمخاطر التعبير التفاضلي وRNA-Seq، يتم تكرار الملاحظات المهمة باستخدام (1) طريقة متعامدة في نفس العينات (مثل PCR في الوقت الفعلي) أو (2) تجربة أخرى، مسجلة مسبقًا في بعض الأحيان، في مجموعة جديدة. هذا الأخير يساعد على ضمان التعميم ويمكن متابعته عادةً مع التحليل التلوي لجميع الأفواج المجمعة. الطريقة الأكثر شيوعًا للحصول على فهم بيولوجي عالي المستوى للنتائج هي تحليل التخصيب الجيني، على الرغم من أنه يتم أحيانًا استخدام طرق الجينات المرشحة. يحدد إثراء مجموعة الجينات ما إذا كان التداخل بين مجموعتين من الجينات ذا دلالة إحصائية، وفي هذه الحالة يكون التداخل بين الجينات ومجموعات الجينات التي يتم التعبير عنها تفاضليًا من مسارات / قواعد البيانات المعروفة (مثل، الجينات الأنطولوجية، KEGG، الأنثروبولوجيا البشرية للأنماط) أو من التحليلات التكميلية في التحليل الجيني نفس البيانات (مثل شبكات التعبير المشترك). تشمل الأدوات الشائعة لإثراء مجموعة الجينات واجهات الويب (مثل، ENRICHR، g: profiler) وحزم البرامج. عند تقييم نتائج التخصيب، يكون أحد الأساليب البحثية هو البحث أولاً عن إثراء البيولوجيا المعروفة كتحقق من الصحة ومن ثم توسيع النطاق للبحث عن بيولوجيا جديدة.
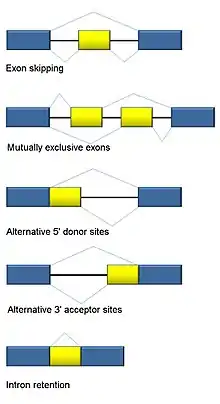
الربط البديل
الربط بين الحمض النووي الريبي جزء لا يتجزأ من حقيقيات النوى ويسهم بشكل كبير في تنظيم البروتين وتنوعه، حيث يحدث في أكثر من 90 ٪ من الجينات البشرية. [99] هناك عدة طرق بديلة للتشكيل: تخطي exon وضع الربط الأكثر شيوعًا في البشر ونواة حقيقيات النوى أعلى، exons حصرية متبادلة، ومتبرع بديل أو مواقع متقبلة، والاحتفاظ بالإنترون (وضع الربط الأكثر شيوعًا في النباتات، والفطريات، والبروتوزوا) الموقع (المروج)، والبوليدينيل البديل. أحد أهداف RNA-Seq هو تحديد أحداث الربط البديلة والاختبار إذا كانت تختلف بين الظروف. تسلسل القراءة الطويلة يلتقط النص الكامل، وبالتالي يقلل من العديد من القضايا في تقدير وفرة isoform، مثل تعيين قراءة غامضة. بالنسبة لقراءة RNA-Seq القصيرة، هناك طرق متعددة لاكتشاف الربط البديل الذي يمكن تصنيفه إلى ثلاث مجموعات رئيسية: [100] [101] [102]
- قائم على العد (أيضًا قائم على الأحداث ، الربط التفاضلي): تقدير استبقاء الإكسون. ومن الأمثلة على ذلك DEXSeq ، [103] MATS ، [104] و SeqGSEA. [105]
- قائم على Isoform (أيضًا وحدات متعددة القراءة ، التعبير التفاضلي isoform) : تقدير الوفرة isoform أولاً ، ثم الوفرة النسبية بين الشروط. الأمثلة على ذلك هي Cufflinks 2 [106] و DiffSplice. [107]
- الختان على أساس Intron: حساب الربط البديل باستخدام تقسيم القراءة. ومن الأمثلة على ذلك MAJIQ [108] و Leafcutter. [102]
يمكن أيضًا استخدام أدوات التعبير الجيني التفاضلي للتعبير التفاضلي isoform إذا تم قياس الأشكال الإسوية مسبقًا مع أدوات أخرى مثل RSEM. [109]
شبكات التعبير (النسخ) المساعد
شبكات التعبير (النسخ) المساعد هي تمثيلات مستمدة من البيانات من الجينات تتصرف بطريقة مماثلة عبر الأنسجة والظروف التجريبية. [110] يكمن هدفهم الرئيسي في توليد الفرضيات وأساليب الذنب من جانب الجمعيات لاستنتاج وظائف الجينات غير المعروفة سابقًا. تم استخدام بيانات RNA-Seq لاستنتاج الجينات المشاركة في مسارات محددة تعتمد على ارتباط بيرسون، سواء في النباتات [111] والثدييات. [112] الميزة الرئيسية لبيانات RNA-Seq في هذا النوع من التحليل عبر منصات ميكروأري هي القدرة على تغطية النص بأكمله، مما يتيح إمكانية كشف تمثيلات أكثر اكتمالا لشبكات تنظيم الجينات. يمكن الكشف عن التنظيم التفريقي لعناصر الشكل العظمية للجنس نفسه واستخدامها للتنبؤ ووظائفها البيولوجية. [113] [114] تم بنجاح استخدام تحليل شبكة التعبير المشترك للجينة الموزونة لتحديد الوحدات النمطية للتعبير المشترك والجينات المحورية داخل العضل على بيانات الحمض النووي الريبي. قد تتوافق وحدات التعبير المشترك مع أنواع الخلايا أو المسارات. يمكن تفسير لوحات الوصل الفائقة المتصلة ببعضها البعض كممثلين لوحدة كل منها. إن eigengene هو عبارة عن مجموع مرجح للتعبير عن جميع الجينات في وحدة نمطية. Eigengenes هي علامات حيوية مفيدة (ميزات) للتشخيص والتشخيص. [115] تم اقتراح مناهج تحويل التباين المستقر لتقدير معاملات الارتباط بناءً على بيانات RNA seq.
اكتشاف البديل
يلتقط RNA-Seq تباين الحمض النووي، بما في ذلك متغيرات النوكليوتيدات الفردية والإدخالات الصغيرة / الحذف. والتباين الهيكلي. يشبه استدعاء المتغير في RNA-Seq استدعاء متغير الحمض النووي، وغالبًا ما يستخدم نفس الأدوات (بما في ذلك SAMtools mpileup [116] و GATK HaplotypeCaller [117] ) مع تعديلات لحساب الربط. أحد الأبعاد الفريدة لمتغيرات الحمض النووي الريبي هو التعبير الخاص بالأليل (ASE) : قد يتم التعبير عن المتغيرات من النمط الفرداني واحد بشكل تفضيلي بسبب التأثيرات التنظيمية بما في ذلك البصمة والتعبير عن موقع السمات الكمية، والمتغيرات النادرة غير المشفرة. [118] [119] تتضمن حدود تحديد متغير الحمض النووي الريبي (RNA) أنه يعكس فقط المناطق المعبر عنها (عند البشر، <5٪ من الجينوم) ولديه جودة أقل مقارنة بتسلسل الحمض النووي المباشر.
تحرير الحمض النووي الريبي (التعديلات بعد النسخ)
يمكن أن يساعد وجود تسلسلات مطابقة للجينوم والنسخ للفرد في الكشف عن التعديلات بعد النسخ ( تحرير RNA ). [3] يتم تحديد حدث تعديل ما بعد النسخ إذا كان نص الجين يحتوي على أليل / متغير لم يلاحظ في البيانات الجينية.

كشف الجينات الانصهار
بسبب التعديلات الهيكلية المختلفة في الجينوم، اكتسبت جينات الاندماج الانتباه بسبب علاقتها بالسرطان. [120] إن قدرة RNA-Seq على تحليل النص الكامل للعينة بطريقة غير متحيزة تجعلها أداة جذابة للعثور على هذه الأنواع من الأحداث الشائعة في السرطان. [4]
وتأتي الفكرة من عملية محاذاة القراءات النصية القصيرة إلى الجينوم المرجعي. ستقع معظم القراءات القصيرة ضمن إكسون واحد كامل، ومن المتوقع أن تحدد مجموعة أصغر ولكن لا تزال كبيرة على تقاطعات إكسون المعروفة. عندئذٍ، سيتم تحليل القراءات القصيرة المتبقية غير المعينة لتحديد ما إذا كانت تتطابق مع تقاطع إكسون حيث يأتي الإكسون من جينات مختلفة. قد يكون هذا دليلًا على حدوث اندماج محتمل، ولكن نظرًا لطول القراءات، فقد يكون هذا مزدحم للغاية. هناك طريقة بديلة تتمثل في استخدام قراءات نهاية الزوج، عندما يقوم عدد كبير يحتمل من القراءات المقترنة بتعيين كل طرف إلى إكسون مختلف، مما يوفر تغطية أفضل لهذه الأحداث ومع ذلك، فإن النتيجة النهائية تتكون من مجموعات متعددة وربما جديدة من الجينات توفر نقطة انطلاق مثالية لمزيد من التحقق من الصحة.
التاريخ
.png.webp)
تم تطوير RNA-Seq لأول مرة في منتصف عام 2000 مع ظهور تقنية التسلسل من الجيل التالي. [121] تتضمن المخطوطات الأولى التي استخدمت RNA-Seq حتى بدون استخدام المصطلح تلك الخاصة بخطوط خلايا سرطان البروستاتا [122] (بتاريخ 2006) ، و Medicago truncatula [123] (2006) ، والذرة [124] (2007) ، و Arabidopsis thaliana [125] (2007) ، في حين أن مصطلح "RNA-Seq" نفسه ذكر لأول مرة في عام 2008. [126] يزداد عدد المخطوطات التي تشير إلى RNA-Seq في العنوان أو الملخص (الشكل ، الخط الأزرق) بشكل مستمر مع 6754 مخطوطة نشرت في عام 2018 ( رابط للبحث في PubMed ). تقاطع RNA-Seq والدواء (الشكل ، الخط الذهبي ، رابط البحث في النشرات الطبية PubMed ) له مرونة مماثلة.
تطبيقات على الطب
RNA-Seq لديه القدرة على تحديد البيولوجيا المرضية الجديدة ، المؤشرات الحيوية الشخصية للمؤشرات السريرية ، استنتاج مسارات قابلة للتخدير ، وجعل التشخيص الوراثي. يمكن تخصيص هذه النتائج بشكل أكبر للمجموعات الفرعية أو حتى للمرضى الأفراد، مما قد يؤدي إلى تسليط الضوء على المزيد من الوقاية الفعالة والتشخيص والعلاج. تملي جدوى هذا النهج جزئياً التكاليف في المال والوقت؛ أحد القيود ذات الصلة هو فريق المتخصصين المطلوب (خبراء المعلومات الحيوية والأطباء / الأطباء والباحثون الأساسيون والفنيون) لتفسير كامل الكم الهائل من البيانات الناتجة عن هذا التحليل.
جهود التسلسل الجيني على نطاق واسع
تم التركيز بشكل كبير على بيانات RNA-Seq بعد أن استخدمت مشاريع موسوعة عناصر الحمض النووي (ENCODE) وأطلس سرطان الجينوم (TCGA) هذا النهج لتوصيف عشرات من خطوط الخلايا [127] وآلاف من عينات الورم الأولية، [128] على التوالي. تهدف ENCODE إلى تحديد المناطق التنظيمية على نطاق الجينوم في مجموعة مختلفة من خطوط الخلايا والبيانات transcriptomic هي الأهم من أجل فهم تأثير المصب لتلك الطبقات التنظيمية الجينية والوراثية. يهدف TCGA، بدلاً من ذلك، إلى جمع وتحليل آلاف من عينات المرضى من 30 نوعًا مختلفًا من الأورام من أجل فهم الآليات الأساسية للتحول والتطور الخبيث. في هذا السياق، توفر بيانات RNA-Seq لقطة فريدة للحالة النصية للمرض وتنظر إلى مجموعة غير متحيزة من النصوص التي تسمح بتحديد النصوص الجديدة ونصوص الاندماج وRNA غير المشفرة التي يمكن اكتشافها باستخدام تقنيات مختلفة.
المراجع
- Shafee, Thomas؛ Lowe, Rohan (2017)، "Eukaryotic and prokaryotic gene structure"، WikiJournal of Medicine (باللغة الإنجليزية)، 4 (1)، doi:10.15347/wjm/2017.002.
- "RNA sequencing: platform selection, experimental design, and data interpretation"، Nucleic Acid Therapeutics، 22 (4): 271–4، أغسطس 2012، doi:10.1089/nat.2012.0367، PMID 22830413.
- "RNA-Seq: a revolutionary tool for transcriptomics"، Nature Reviews. Genetics، 10 (1): 57–63، يناير 2009، doi:10.1038/nrg2484، PMID 19015660.
- "Transcriptome sequencing to detect gene fusions in cancer"، Nature، 458 (7234): 97–101، مارس 2009، Bibcode:2009Natur.458...97M، doi:10.1038/nature07638، PMID 19136943.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments"، Nature Protocols، 7 (8): 1534–50، يوليو 2012، doi:10.1038/nprot.2012.086، PMID 22836135.
- "Highly multiplexed subcellular RNA sequencing in situ"، Science، 343 (6177): 1360–3، مارس 2014، Bibcode:2014Sci...343.1360L، doi:10.1126/science.1250212، PMID 24578530.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "RNA Sequencing and Analysis"، Cold Spring Harbor Protocols، 2015 (11): 951–69، أبريل 2015، doi:10.1101/pdb.top084970، PMID 25870306.
- "Informatics for RNA Sequencing: A Web Resource for Analysis on the Cloud"، PLoS Computational Biology، 11 (8): e1004393، أغسطس 2015، Bibcode:2015PLSCB..11E4393G، doi:10.1371/journal.pcbi.1004393، PMID 26248053.
- "RNA-seqlopedia"، rnaseq.uoregon.edu، مؤرشف من الأصل في 30 ديسمبر 2019، اطلع عليه بتاريخ 08 فبراير 2017.
- "Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing"، BioTechniques، 45 (1): 81–94، يوليو 2008، doi:10.2144/000112900، PMID 18611170، مؤرشف من الأصل في 31 مارس 2019.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Mapping and quantifying mammalian transcriptomes by RNA-Seq"، Nature Methods، 5 (7): 621–8، يوليو 2008، doi:10.1038/nmeth.1226، PMID 18516045.
- "Effect of RNA integrity on uniquely mapped reads in RNA-Seq"، BMC Research Notes، 7 (1): 753، أكتوبر 2014، doi:10.1186/1756-0500-7-753، PMID 25339126.
- "Quantitative comparison of EST libraries requires compensation for systematic biases in cDNA generation"، BMC Bioinformatics، 7: 77، فبراير 2006، doi:10.1186/1471-2105-7-77، PMID 16503995.
- "Highly parallel direct RNA sequencing on an array of nanopores"، Nature Methods، 15 (3): 201–206، مارس 2018، doi:10.1038/nmeth.4577، PMID 29334379.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - ""Single-cell sequencing-based technologies will revolutionize whole-organism science"، Nature Reviews. Genetics، 14 (9): 618–30، سبتمبر 2013، doi:10.1038/nrg3542، PMID 23897237."
- "The technology and biology of single-cell RNA sequencing"، Molecular Cell، 58 (4): 610–20، مايو 2015، doi:10.1016/j.molcel.2015.04.005، PMID 26000846.
- "A revised airway epithelial hierarchy includes CFTR-expressing ionocytes"، Nature، 560 (7718): 319–324، أغسطس 2018، Bibcode:2018Natur.560..319M، doi:10.1038/s41586-018-0393-7، PMID 30069044.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte"، Nature، 560 (7718): 377–381، أغسطس 2018، Bibcode:2018Natur.560..377P، doi:10.1038/s41586-018-0394-6، PMID 30069046.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells"، Cell، 161 (5): 1187–1201، مايو 2015، doi:10.1016/j.cell.2015.04.044، PMID 26000487.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets"، Cell، 161 (5): 1202–1214، مايو 2015، doi:10.1016/j.cell.2015.05.002، PMID 26000488.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - ""Methods, Challenges and Potentials of Single Cell RNA-seq"، Biology، 1 (3): 658–67، نوفمبر 2012، doi:10.3390/biology1030658، PMID 24832513."
- "The promise of single-cell sequencing"، Nature Methods، 11 (1): 25–7، يناير 2014، doi:10.1038/nmeth.2769، PMID 24524134.
- "mRNA-Seq whole-transcriptome analysis of a single cell"، Nature Methods، 6 (5): 377–82، مايو 2009، doi:10.1038/NMETH.1315، PMID 19349980.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq"، Genome Research، 21 (7): 1160–7، يوليو 2011، doi:10.1101/gr.110882.110، PMID 21543516.
- "Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells"، Nature Biotechnology، 30 (8): 777–82، أغسطس 2012، doi:10.1038/nbt.2282، PMID 22820318.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification"، Cell Reports، 2 (3): 666–73، سبتمبر 2012، doi:10.1016/j.celrep.2012.08.003، PMID 22939981.
- "High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes"، bioRxiv، 2018، doi:10.1101/424945.
- "Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity"، Genome Biology، 14 (4): R31، أبريل 2013، doi:10.1186/gb-2013-14-4-r31، PMID 23594475.
- "C1 CAGE detects transcription start sites and enhancer activity at single-cell resolution"، Nature Communications، 10 (1): 360، يناير 2019، Bibcode:2019NatCo..10..360K، doi:10.1038/s41467-018-08126-5، PMID 30664627.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "How to design a single-cell RNA-sequencing experiment: pitfalls, challenges and perspectives"، Briefings in Bioinformatics: bby007، يناير 2018، doi:10.1093/bib/bby007، PMID 29394315.
- "Multiplexed quantification of proteins and transcripts in single cells"، Nature Biotechnology، 35 (10): 936–939، أكتوبر 2017، doi:10.1038/nbt.3973، PMID 28854175.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Simultaneous epitope and transcriptome measurement in single cells"، Nature Methods، 14 (9): 865–868، سبتمبر 2017، doi:10.1038/nmeth.4380، PMID 28759029.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain"، Nature Biotechnology، 36 (5): 442–450، يونيو 2018، doi:10.1038/nbt.4103، PMID 29608178.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Circulating tumour cell (CTC) counts as intermediate end points in castration-resistant prostate cancer (CRPC): a single-centre experience"، Annals of Oncology، 20 (1): 27–33، يناير 2009، doi:10.1093/annonc/mdn544، PMID 18695026.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Single-Cell Transcriptomic Analysis of Tumor Heterogeneity"، Trends in Cancer (باللغة الإنجليزية)، 4 (4): 264–268، أبريل 2018، doi:10.1016/j.trecan.2018.02.003، PMID 29606308، مؤرشف من الأصل في 11 مايو 2020.
- "A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade"، Cell (باللغة الإنجليزية)، 175 (4): 984–997.e24، نوفمبر 2018، doi:10.1016/j.cell.2018.09.006، PMID 30388455، مؤرشف من الأصل في 11 مايو 2020.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Single-cell RNA-seq of rheumatoid arthritis synovial tissue using low-cost microfluidic instrumentation"، Nature Communications، 9 (1): 791، فبراير 2018، Bibcode:2018NatCo...9..791S، doi:10.1038/s41467-017-02659-x، PMID 29476078.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Pathogen Cell-to-Cell Variability Drives Heterogeneity in Host Immune Responses"، Cell، 162 (6): 1309–21، سبتمبر 2015، doi:10.1016/j.cell.2015.08.027، PMID 26343579.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Comprehensive single-cell transcriptional profiling of a multicellular organism"، Science، 357 (6352): 661–667، أغسطس 2017، Bibcode:2017Sci...357..661C، doi:10.1126/science.aam8940، PMID 28818938.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics"، Science، 360 (6391): eaaq1723، مايو 2018، doi:10.1126/science.aaq1723، PMID 29674432، مؤرشف من الأصل في 11 مايو 2020.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Schmidtea mediterranea"، Science، 360 (6391): eaaq1736، مايو 2018، doi:10.1126/science.aaq1736، PMID 29674431.
- "Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo"، Science، 360 (6392): 981–987، يونيو 2018، Bibcode:2018Sci...360..981W، doi:10.1126/science.aar4362، PMID 29700229.
- "Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis"، Science، 360 (6392): eaar3131، يونيو 2018، doi:10.1126/science.aar3131، PMID 29700225.
- "The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution"، Science، 360 (6392): eaar5780، يونيو 2018، doi:10.1126/science.aar5780، PMID 29700227.
- You, Jia، "Science's 2018 Breakthrough of the Year: tracking development cell by cell"، Science Magazine، American Association for the Advancement of Science، مؤرشف من الأصل في 06 ديسمبر 2019.
- "Determination of tag density required for digital transcriptome analysis: application to an androgen-sensitive prostate cancer model"، Proceedings of the National Academy of Sciences of the United States of America، 105 (51): 20179–84، ديسمبر 2008، Bibcode:2008PNAS..10520179L، doi:10.1073/pnas.0807121105، PMID 19088194.
- "Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses"، Nature Protocols، 7 (3): 500–7، فبراير 2012، doi:10.1038/nprot.2011.457، PMID 22343431.
- "Reference-based compression of short-read sequences using path encoding"، Bioinformatics، 31 (12): 1920–8، يونيو 2015، doi:10.1093/bioinformatics/btv071، PMID 25649622.
- "Full-length transcriptome assembly from RNA-Seq data without a reference genome"، Nature Biotechnology، 29 (7): 644–52، مايو 2011، doi:10.1038/nbt.1883، PMID 21572440.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "De Novo Assembly Using Illumina Reads" (PDF)، مؤرشف من الأصل (PDF) في 30 أبريل 2019، اطلع عليه بتاريخ 22 أكتوبر 2016.
- "Velvet: algorithms for de novo short read assembly using de Bruijn graphs"، Genome Research، 18 (5): 821–9، مايو 2008، doi:10.1101/gr.074492.107، PMID 18349386.
- Oases: a transcriptome assembler for very short reads نسخة محفوظة 29 نوفمبر 2018 على موقع واي باك مشين.
- "Bridger: a new framework for de novo transcriptome assembly using RNA-seq data"، Genome Biology، 16 (1): 30، فبراير 2015، doi:10.1186/s13059-015-0596-2، PMID 25723335.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Evaluation of de novo transcriptome assemblies from RNA-Seq data"، Genome Biology، 15 (12): 553، ديسمبر 2014، doi:10.1186/s13059-014-0553-5، PMID 25608678.
- "STAR: ultrafast universal RNA-seq aligner"، Bioinformatics، 29 (1): 15–21، يناير 2013، doi:10.1093/bioinformatics/bts635، PMID 23104886.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Ultrafast and memory-efficient alignment of short DNA sequences to the human genome"، Genome Biology، 10 (3): R25، 2009، doi:10.1186/gb-2009-10-3-r25، PMID 19261174.
- "TopHat: discovering splice junctions with RNA-Seq"، Bioinformatics، 25 (9): 1105–11، مايو 2009، doi:10.1093/bioinformatics/btp120، PMID 19289445.
- "Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks"، Nature Protocols، 7 (3): 562–78، مارس 2012، doi:10.1038/nprot.2012.016، PMID 22383036.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote"، Nucleic Acids Research، 41 (10): e108، مايو 2013، doi:10.1093/nar/gkt214، PMID 23558742.
- "HISAT: a fast spliced aligner with low memory requirements"، Nature Methods، 12 (4): 357–60، أبريل 2015، doi:10.1038/nmeth.3317، PMID 25751142.
- "Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms"، Nature Biotechnology، 32 (5): 462–4، مايو 2014، arXiv:1308.3700، doi:10.1038/nbt.2862، PMID 24752080.
- "Near-optimal probabilistic RNA-seq quantification"، Nature Biotechnology، 34 (5): 525–7، مايو 2016، doi:10.1038/nbt.3519، PMID 27043002.
- "GMAP: a genomic mapping and alignment program for mRNA and EST sequences"، Bioinformatics، 21 (9): 1859–75، مايو 2005، doi:10.1093/bioinformatics/bti310، PMID 15728110.
- "Simulation-based comprehensive benchmarking of RNA-seq aligners"، Nature Methods (باللغة الإنجليزية)، 14 (2): 135–139، فبراير 2017، doi:10.1038/nmeth.4106، PMID 27941783.
- "Systematic evaluation of spliced alignment programs for RNA-seq data"، Nature Methods (باللغة الإنجليزية)، 10 (12): 1185–91، ديسمبر 2013، doi:10.1038/nmeth.2722، PMID 24185836.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Comparative study of de novo assembly and genome-guided assembly strategies for transcriptome reconstruction based on RNA-Seq"، Science China Life Sciences، 56 (2): 143–55، فبراير 2013، doi:10.1007/s11427-013-4442-z، PMID 23393030.
- "Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species"، GigaScience، 2 (1): 10، يوليو 2013، arXiv:1301.5406، Bibcode:2013arXiv1301.5406B، doi:10.1186/2047-217X-2-10، PMID 23870653.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Comparing protein abundance and mRNA expression levels on a genomic scale"، Genome Biology، 4 (9): 117، 2003، doi:10.1186/gb-2003-4-9-117، PMID 12952525.
- "A comparative study of techniques for differential expression analysis on RNA-Seq data"، PLOS ONE، 9 (8): e103207، أغسطس 2014، Bibcode:2014PLoSO...9j3207Z، doi:10.1371/journal.pone.0103207، PMID 25119138.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "HTSeq--a Python framework to work with high-throughput sequencing data"، Bioinformatics، 31 (2): 166–9، يناير 2015، doi:10.1093/bioinformatics/btu638، PMID 25260700.
- "featureCounts: an efficient general purpose program for assigning sequence reads to genomic features"، Bioinformatics، 30 (7): 923–30، أبريل 2014، arXiv:1305.3347، doi:10.1093/bioinformatics/btt656، PMID 24227677.
- "Rcount: simple and flexible RNA-Seq read counting"، Bioinformatics، 31 (3): 436–7، فبراير 2015، doi:10.1093/bioinformatics/btu680، PMID 25322836.
- "Reducing bias in RNA sequencing data: a novel approach to compute counts"، BMC Bioinformatics، 15 Suppl 1 (Suppl 1): S7، 2014، doi:10.1186/1471-2105-15-s1-s7، PMID 24564404.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Universal count correction for high-throughput sequencing"، PLoS Computational Biology، 10 (3): e1003494، مارس 2014، Bibcode:2014PLSCB..10E3494H، doi:10.1371/journal.pcbi.1003494، PMID 24603409.
- "A scaling normalization method for differential expression analysis of RNA-seq data"، Genome Biology، 11 (3): R25، 2010، doi:10.1186/gb-2010-11-3-r25، PMID 20196867.
- "Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation"، Nature Biotechnology، 28 (5): 511–5، مايو 2010، doi:10.1038/nbt.1621، PMID 20436464.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "What the FPKM? A review of RNA-Seq expression units"، The farrago، 08 مايو 2014، مؤرشف من الأصل في 30 ديسمبر 2019، اطلع عليه بتاريخ 28 مارس 2018.
- "Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples"، Theory in Biosciences = Theorie in den Biowissenschaften، 131 (4): 281–5، ديسمبر 2012، doi:10.1007/s12064-012-0162-3، PMID 22872506.
- "voom: Precision weights unlock linear model analysis tools for RNA-seq read counts"، Genome Biology، 15 (2): R29، فبراير 2014، doi:10.1186/gb-2014-15-2-r29، PMID 24485249.
- "Differential expression analysis for sequence count data"، Genome Biology، 11 (10): R106، 2010، doi:10.1186/gb-2010-11-10-r106، PMID 20979621.
- "edgeR: a Bioconductor package for differential expression analysis of digital gene expression data"، Bioinformatics، 26 (1): 139–40، يناير 2010، doi:10.1093/bioinformatics/btp616، PMID 19910308.
- "Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells"، Cell، 151 (3): 671–83، أكتوبر 2012، doi:10.1016/j.cell.2012.09.019، PMID 23101633.
- "Measuring Absolute RNA Copy Numbers at High Temporal Resolution Reveals Transcriptome Kinetics in Development"، Cell Reports، 14 (3): 632–647، يناير 2016، doi:10.1016/j.celrep.2015.12.050، PMID 26774488.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "limma powers differential expression analyses for RNA-sequencing and microarray studies"، Nucleic Acids Research، 43 (7): e47، أبريل 2015، doi:10.1093/nar/gkv007، PMID 25605792.
- "Bioconductor - Open source software for bioinformatics"، مؤرشف من الأصل في 29 ديسمبر 2019.
- "Orchestrating high-throughput genomic analysis with Bioconductor"، Nature Methods، 12 (2): 115–21، فبراير 2015، doi:10.1038/nmeth.3252، PMID 25633503.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Capturing heterogeneity in gene expression studies by surrogate variable analysis"، PLoS Genetics، 3 (9): 1724–35، سبتمبر 2007، doi:10.1371/journal.pgen.0030161، PMID 17907809.
- "Differential analysis of gene regulation at transcript resolution with RNA-seq" (PDF)، Nature Biotechnology، 31 (1): 46–53، يناير 2013، doi:10.1038/nbt.2450، PMID 23222703، مؤرشف من الأصل (PDF) في 11 مايو 2020.
- "Differential analysis of RNA-seq incorporating quantification uncertainty"، Nature Methods، 14 (7): 687–690، يوليو 2017، doi:10.1038/nmeth.4324، PMID 28581496.
- "Ballgown bridges the gap between transcriptome assembly and expression analysis"، Nature Biotechnology، 33 (3): 243–6، مارس 2015، doi:10.1038/nbt.3172، PMID 25748911.
- "Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis"، Nature Communications، 8 (1): 59، يوليو 2017، Bibcode:2017NatCo...8...59S، doi:10.1038/s41467-017-00050-4، PMID 28680106.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Gene name errors are widespread in the scientific literature"، Genome Biology، 17 (1): 177، أغسطس 2016، doi:10.1186/s13059-016-1044-7، PMID 27552985.
- "A comparison of methods for differential expression analysis of RNA-seq data"، BMC Bioinformatics، 14: 91، مارس 2013، doi:10.1186/1471-2105-14-91، PMID 23497356.
- "RNA-Seq gene profiling--a systematic empirical comparison"، PLOS ONE، 9 (9): e107026، 30 سبتمبر 2014، Bibcode:2014PLoSO...9j7026F، doi:10.1371/journal.pone.0107026، PMID 25268973.
- "Comparison of software packages for detecting differential expression in RNA-seq studies"، Briefings in Bioinformatics، 16 (1): 59–70، يناير 2015، doi:10.1093/bib/bbt086، PMID 24300110.
- "Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data"، Genome Biology، 14 (9): R95، 2013، doi:10.1186/gb-2013-14-9-r95، PMID 24020486.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "A survey of best practices for RNA-seq data analysis"، Genome Biology، 17 (1): 13، يناير 2016، doi:10.1186/s13059-016-0881-8، PMID 26813401.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "RNA-Seq differential expression analysis: An extended review and a software tool"، PLOS ONE، 12 (12): e0190152، 21 ديسمبر 2017، Bibcode:2017PLoSO..1290152C، doi:10.1371/journal.pone.0190152، PMID 29267363.
- "Alternative splicing and evolution: diversification, exon definition and function"، Nature Reviews. Genetics، 11 (5): 345–55، مايو 2010، doi:10.1038/nrg2776، PMID 20376054.
- "Comparisons of computational methods for differential alternative splicing detection using RNA-seq in plant systems"، BMC Bioinformatics، 15 (1): 364، ديسمبر 2014، doi:10.1186/s12859-014-0364-4، PMID 25511303.
- Pachter, Lior (19 أبريل 2011)، "Models for transcript quantification from RNA-Seq" (باللغة الإنجليزية)، arXiv:1104.3889، Bibcode:2011arXiv1104.3889P.
{{استشهاد بدورية محكمة}}: Cite journal requires|journal=(مساعدة) - "Annotation-free quantification of RNA splicing using LeafCutter"، Nature Genetics، 50 (1): 151–158، يناير 2018، doi:10.1038/s41588-017-0004-9، PMID 29229983.
- "Detecting differential usage of exons from RNA-seq data"، Genome Research، 22 (10): 2008–17، أكتوبر 2012، doi:10.1101/gr.133744.111، PMID 22722343.
- "MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data"، Nucleic Acids Research، 40 (8): e61، أبريل 2012، doi:10.1093/nar/gkr1291، PMID 22266656.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "SeqGSEA: a Bioconductor package for gene set enrichment analysis of RNA-Seq data integrating differential expression and splicing"، Bioinformatics، 30 (12): 1777–9، يونيو 2014، doi:10.1093/bioinformatics/btu090، PMID 24535097.
- "Differential analysis of gene regulation at transcript resolution with RNA-seq"، Nature Biotechnology، 31 (1): 46–53، يناير 2013، doi:10.1038/nbt.2450، PMID 23222703.
- "DiffSplice: the genome-wide detection of differential splicing events with RNA-seq"، Nucleic Acids Research، 41 (2): e39، يناير 2013، doi:10.1093/nar/gks1026، PMID 23155066.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "A new view of transcriptome complexity and regulation through the lens of local splicing variations"، eLife، 5: e11752، فبراير 2016، doi:10.7554/eLife.11752، PMID 26829591.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "A benchmarking of workflows for detecting differential splicing and differential expression at isoform level in human RNA-seq studies"، Briefings in Bioinformatics، 20 (2): 471–481، مارس 2019، doi:10.1093/bib/bbx122، PMID 29040385.
- "A combined algorithm for genome-wide prediction of protein function"، Nature، 402 (6757): 83–6، نوفمبر 1999، Bibcode:1999Natur.402...83M، doi:10.1038/47048، PMID 10573421.
- "Comparative study of RNA-seq- and microarray-derived coexpression networks in Arabidopsis thaliana"، Bioinformatics، 29 (6): 717–24، مارس 2013، doi:10.1093/bioinformatics/btt053، PMID 23376351.
- "Utilizing RNA-Seq data for de novo coexpression network inference"، Bioinformatics، 28 (12): 1592–7، يونيو 2012، doi:10.1093/bioinformatics/bts245، PMID 22556371.
- "Systematically differentiating functions for alternatively spliced isoforms through integrating RNA-seq data"، PLoS Computational Biology، 9 (11): e1003314، نوفمبر 2013، Bibcode:2013PLSCB...9E3314E، doi:10.1371/journal.pcbi.1003314، PMID 24244129.
- "The emerging era of genomic data integration for analyzing splice isoform function"، Trends in Genetics، 30 (8): 340–7، أغسطس 2014، doi:10.1016/j.tig.2014.05.005، PMID 24951248.
- "Large-scale gene network analysis reveals the significance of extracellular matrix pathway and homeobox genes in acute myeloid leukemia: an introduction to the Pigengene package and its applications"، BMC Medical Genomics، 10 (1): 16، مارس 2017، doi:10.1186/s12920-017-0253-6، PMID 28298217.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "The Sequence Alignment/Map format and SAMtools"، Bioinformatics، 25 (16): 2078–9، أغسطس 2009، doi:10.1093/bioinformatics/btp352، PMID 19505943.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "A framework for variation discovery and genotyping using next-generation DNA sequencing data"، Nature Genetics، 43 (5): 491–8، مايو 2011، doi:10.1038/ng.806، PMID 21478889.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Genetic effects on gene expression across human tissues"، Nature، 550 (7675): 204–213، أكتوبر 2017، Bibcode:2017Natur.550..204A، doi:10.1038/nature24277، PMID 29022597.
- "ORE Identifies Extreme Expression Effects Enriched for Rare Variants"، Bioinformatics، مارس 2019، doi:10.1093/bioinformatics/btz202، PMID 30903145.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Recurrent fusion oncogenes in carcinomas"، Critical Reviews in Oncogenesis، 12 (3–4): 257–71، ديسمبر 2006، doi:10.1615/critrevoncog.v12.i3-4.40، PMID 17425505.
- "Discovering New Biology through Sequencing of RNA"، Plant Physiology، 169 (3): 1524–31، نوفمبر 2015، doi:10.1104/pp.15.01081، PMID 26353759.
- "Analysis of the prostate cancer cell line LNCaP transcriptome using a sequencing-by-synthesis approach"، BMC Genomics، 7: 246، سبتمبر 2006، doi:10.1186/1471-2164-7-246، PMID 17010196.
{{استشهاد بدورية محكمة}}: الوسيط|displayauthors=6غير صالح (مساعدة) - "Sequencing Medicago truncatula expressed sequenced tags using 454 Life Sciences technology"، BMC Genomics، 7: 272، أكتوبر 2006، doi:10.1186/1471-2164-7-272، PMID 17062153.
- "Gene discovery and annotation using LCM-454 transcriptome sequencing"، Genome Research، 17 (1): 69–73، يناير 2007، doi:10.1101/gr.5145806، PMID 17095711.
- "Sampling the Arabidopsis transcriptome with massively parallel pyrosequencing"، Plant Physiology، 144 (1): 32–42، مايو 2007، doi:10.1104/pp.107.096677، PMID 17351049.
- "The transcriptional landscape of the yeast genome defined by RNA sequencing"، Science، 320 (5881): 1344–9، يونيو 2008، Bibcode:2008Sci...320.1344N، doi:10.1126/science.1158441، PMID 18451266.
- "ENCODE Data Matrix"، مؤرشف من الأصل في 30 مايو 2019، اطلع عليه بتاريخ 28 يوليو 2013.
- "The Cancer Genome Atlas - Data Portal"، مؤرشف من الأصل في 16 أبريل 2016، اطلع عليه بتاريخ 28 يوليو 2013.
 بوابة علم الأحياء الخلوي والجزيئي
بوابة علم الأحياء الخلوي والجزيئي