عوز أنزيم ميفالونات كيناز
عوز أنزيم ميفالونات كيناز (بالإنجليزية: Mevalonate kinase deficiency) هو اضطراب أيضي يُورث بشكل جسدي متنح، تتعطل فيه عملية استقلاب الكوليسترول وبعض أنواع الكربوهيدرات، ويتميز بمستويات عالية من الغلوبولين المناعي (دي) في الدم.[1][2]
| عوز أنزيم ميفالونات كيناز | |
|---|---|
| Mevalonate kinase deficiency | |
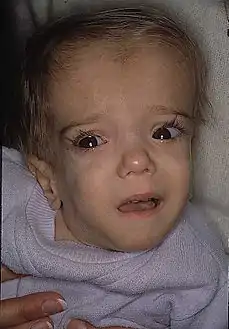 طفل عمره 21 شهر مصاب بعوز أنزيم ميفالونات كيناز تظهر عليه مظاهر تشوهات وجهية قحفية طفل عمره 21 شهر مصاب بعوز أنزيم ميفالونات كيناز تظهر عليه مظاهر تشوهات وجهية قحفية | |
| معلومات عامة | |
| الاختصاص | علم الدم، وطب الجهاز العصبي، وعلم المناعة، وعلم الوراثة الطبية، وعلم الغدد الصم |
| من أنواع | مرض بيروكسيزومي ، واضطراب جيني |

يساهم أنزيم ميفالونات كيناز في الاستقلاب الحيوي للكوليسترول والأيزوبرنويد، وهو ضروري لتحويل الميفالونيت إلى ميفالونيت 5 فوسفات. يتسبب نقص ميفالونات كيناز في تراكم الميفالونيت في البول، وهو الأمر الذي يؤدي لتقليل نشاط الإنزيم مرة أخرى. وصف هذا الاضطراب لأول مرة باسم HIDS في عام 1984.[3]
الوراثة
يُورث اضطراب عوز ميفالونات كيناز بطريقة جسمية متنحية، وهذا يعني أنه يجب على الطفل أن يرث الجين المعيب من كلا الوالدين حتى يصاب بالمرض، وهو نموذج على طفرة فقدان الوظيفة.[4]
الغلوبولين المناعي دي
ينتج الغلوبولين المناعي دي من نوع معين من خلايا الدم البيضاء. هناك خمس فئات من الغلوبولين المناعي تلعب جميعها دوراً هاماً في مناعة الجسم، ولاتزال وظيفة الغلوبولين المناعي دي غير واضحة بدقة حتى الآن.
متلازمة فرط الغلوبولين المناعي دي
تتميز متلازمة فرط الغلوبولين المناعي دي بنوبات من الحمى المتكررة، وهي متلازمة حمى دورية وصفها الطبيب خوسيه فان دير مير في عام 1984،[5] ثم وثقت في سجلات المركز الطبي بجامعة لايدن، وحتى الآن لم يشخص أكثر من 300 حالة في جميع أنحاء العالم.[6]
العلامات والأعراض
تعتبر متلازمة عوز ميفالونات كيناز واحدةً من متلازمات الحمى الدورية. تتميز بنوبات من الحمى والألم المفصلي وآفات جلدية متعددة مثل التقرحات الفموية والإسهال. تظهر التحاليل المخبرية ارتفاعاً حاداً في مستويات الغلوبولين المناعي دي وسرعة التثفل والبروتين الارتكاسي. وصفت حالات متلازمة عوز ميفالونات كيناز بشكل أساسي في هولندا وفرنسا، على الرغم من أن السجل الدولي يتضمن عدداً من الحالات من بلدان أخرى.[7]
يشمل التشخيص التفريقي: الحمى مجهولة السبب، وحمى البحر الأبيض المتوسط العائلية.[7]
الأسباب
يعاني كل الناس المصابين بمتلازمة نقص ميفالونات كيناز من طفرات في الجين المرتبط بهذا الأنزيم،[8][9] وقد وصفت نوبات مماثلة من الحمى عند مرضى يعانون من بيلة حمض الميفالونيك، وهو خلل ولادي في عملية الاستقلاب ينظر إليه اليوم على أنه شكل حاد من متلازمة نقص ميفالونات كيناز.[7]
الفيزيولوجيا المرضية
لا يعرف حتى الآن كيف تسبب طفرات ميفالونات كيناز نوبات الحمى، ولكن يُفترض أنَّ المنتجات الأخرى الناتجة عن مسار التخليق الحيوي للكوليسترول قد تلعب دوراً في ذلك.[7]
التشخيص
يؤدي عوز ميفالونات كيناز إلى تراكم حمض الميفالونيك في البول، ويمكن تأكيد تشخيص الإصابة بهذا المرض عن طريق معايرة نسبة حمض الميفالونيك في البول.[10] وُصف هذا الاضطراب لأول مرة في عام 1985،[11] وهو يُصنف على أنه خطأ أيضي ولادي، وعادة ما يؤدي إلى تأخر في النمو ونقص التوتر وفقر الدم وتضخم الكبد والطحال والعديد من مظاهر التشوه والتخلف العقلي وفشل النمو.
العلاج
لا يوجد علاج لمتلازمة نقص ميفالونات كيناز، ولكن يمكن الحد من الالتهابات والأعراض الأخرى إلى حد ما، يمكن استخدام الانترلوكين 1 الذي ثبت أنه يقلل العملية الالتهابية السريرية والكيميائية، وهو الأمر الذي ثبت أنه ينقص بشكل فعال من وتيرة وشدة الهجمات الالتهابية عند استخدامه بشكلٍ يومي، لكن من مساوئ استخدام هذا الدواء حدوث ألم في موقع الحقن، وعودة نوبات الحرارة بعد إيقاف الدواء بفترة وجيزة.
من الأدوية المستخدمة أيضاً «كاناكينوماب» وهو عبارة عن جسم مضاد وحيد النسيلة طويل المفعول، وقد أثبت أنه فعال في تقليل كل من تكرار النوبات وشدتها في المرضى الذين يعانون من متلازمة نقص الميفالونات الخفيفة والشديدة، وقد أثبتت الدراسات أنه يقلل من التأثيرات الفيزيولوجية، ولكن التحاليل المخبرية الكيميائية الحيوية تبقى مرتفعة.
يستفيد معظم المرضى من العلاجات المختلفة المضادة للإنترلوكين 1، ولكن في بعض الحالات يكون المرض مُعنِّداً على هذه العلاجات، وتقترح الدراسات في هذه الحالة استخدام الأدوية المضادة للإنترلوكين 6، ففي دراسة أجريت على شابة مصابة بالمتلازمة أدى استخدام هذه الأدوية لتقليل شدة وعدد النوبات الالتهابية، على الرغم من استمرار وجود الأعراض بشكل خفيف.
أثبتت بعض الدراسات الحديثة تأثيراً مفيداً لزراعة الخلايا الجذعية المكوِّنة للدم في حالات نقص ميفالونات كيناز الحاد، حيث حدث تحسن في المادة الدماغية على التصوير بالرنين المغناطيسي بعد زرع هذه الخلايا الجذعية، أما زرع الكبد فلم يثبت أنَّ له تأثيرات إيجابية لعلاج هذا المرض.
مراجع
- "Mevalonate kinase deficiency"، مؤرشف من الأصل في 12 سبتمبر 2018.
- Mancini J, Philip N, Chabrol B, Divry P, Rolland MO, Pinsard N (مايو–يونيو 1993)، "Mevalonic aciduria in 3 siblings: a new recognizable metabolic encephalopathy"، Pediatr. Neurol.، 9 (3): 243–246، doi:10.1016/0887-8994(93)90095-T، PMID 8352861.
- Background information, HIDS.net, retrieved 28 August 2016 نسخة محفوظة 1 أبريل 2019 على موقع واي باك مشين.
- "Mevalonate Kinase Deficiency"، مؤرشف من الأصل في 22 أكتوبر 2018.
- van der Meer JW, Vossen JM, Radl J, وآخرون (مايو 1984)، "Hyperimmunoglobulinaemia D and periodic fever: a new syndrome"، Lancet، 1 (8386): 1087–90، doi:10.1016/S0140-6736(84)92505-4، PMID 6144826.
- "OMIM Entry - * 251170 - MEVALONATE KINASE; MVK"، omim.org، مؤرشف من الأصل في 13 سبتمبر 2019.
- Drenth JP, van der Meer JW (ديسمبر 2001)، "Hereditary periodic fever"، N. Engl. J. Med.، 345 (24): 1748–57، doi:10.1056/NEJMra010200، PMID 11742050.
- Drenth JP, Cuisset L, Grateau G, وآخرون (يونيو 1999)، "Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group"، Nat. Genet.، 22 (2): 178–81، doi:10.1038/9696، PMID 10369262.
- Houten SM, Kuis W, Duran M, وآخرون (يونيو 1999)، "Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome"، Nat. Genet.، 22 (2): 175–7، doi:10.1038/9691، PMID 10369261.
- Bretón Martínez JR, Cánovas Martínez A, Casaña Pérez S, Escribá Alepuz J, Giménez Vázquez F (أكتوبر 2007)، "Mevalonic aciduria: report of two cases"، J. Inherit. Metab. Dis.، 30 (5): 829، doi:10.1007/s10545-007-0618-7، PMID 17578678.
- Berger R, Smit GP, Schierbeek H, Bijsterveld K, le Coultre R (أكتوبر 1985)، "Mevalonic aciduria: an inborn error of cholesterol biosynthesis?"، Clin. Chim. Acta، 152 (1–2): 219–222، doi:10.1016/0009-8981(85)90195-0، PMID 4053401.
 بوابة طب
بوابة طب