فسفوجلوكوميوتاز (إنزيم)
فسفوجلوكوموتاز ( ر.ت.إ 5.4.2.2 ) هو إنزيم ينقل مجموعة الفوسفات على مونومر الجلوكوز α-D- من الموضع 1 'إلى 6' في الاتجاه الأمامي أو 6 'إلى 1' في الاتجاه العكسي. بأكثر دقة، انه يسهل عملية التحويل المتبادل بين الجلوكوز 1- فوسفات و الجلوكوز6 فوسفات.
الوظيفة البيولوجية
دور الانزيم في تحلل الجليكوجين
بعد أن يحفز فوسفوريلاز الجليكوجين الانقسام الفسفوري لمخلفات الجلوكوزيل من بوليمر الجليكوجين، يحتوي الجلوكوز المحرر على مجموعة فوسفات على الكربون 1. إن جزيءالجلوكوز 1-فوسفات ليس في حد ذاته وسيطًا استقلابيًا مفيدًا، ولكن الفوسفوجلوكوموتاز يحفز تحويل هذا الجلوكوز 1-فوسفات إلى الجلوكوز 6-فوسفات (انظر أدناه للتعرف على آلية هذا التفاعل).
يعتمد مصير التمثيل الغذائي للجلوكوز 6 فوسفات على احتياجات الخلية في الوقت الذي يتم إنشاؤه فيه. إذا كانت الخلية منخفضة الطاقة، فسوف يسافر الجلوكوز 6 فوسفات في المسار الجليكوليتي، مما يؤدي في النهاية إلى جزيئين من الأدينوزين ثلاثي الفوسفات. إذا كانت الخلية بحاجة إلى وسيطات صناعية حيوية، فسوف يدخل الجلوكوز 6 فوسفات إلى مسار فوسفات البنتوز، حيث سيخضع لسلسلة من التفاعلات للحصول على الريبوز و / أو NADPH، اعتمادًا على الظروف الخلوية.
إذا كان تحلل الجليكوجين يحدث في الكبد، يمكن تحويل الجلوكوز 6 فوسفات إلى الجلوكوز بواسطة إنزيم الجلوكوز 6-فوسفاتاز. ثم يتم تحرير الجلوكوز المنتج في الكبد إلى مجرى الدم لاستخدامه في الأعضاء الأخرى. على عكس ذلك، لا تحتوي خلايا العضلات على إنزيم الجلوكوز 6 -فوسفاتاز، لذا لا يمكنها مشاركة مخازن الجليكوجين مع بقية الجسم.
دور الإنزيم في تكوين السكر
يعمل فوسفوجلوكوموتاز أيضًا بطريقة عكسية عندما تكون مستويات الجلوكوز في الدم مرتفعة. في هذه الحالة، يحفز فوسفوجلوكوموتاز تحويل الجلوكوز 6-فوسفات (الذي يتم إنشاؤه بسهولة من الجلوكوز بفعل انزيم الهيكسوكيناز ) إلى الجلوكوز 1-فوسفات.
يمكن أن يتفاعل هذا الجلوكوز 1-فوسفات بعد ذلك مع UTP لإنتاج UDP-glucose في تفاعل محفز بواسطة UDP-glucose-pyrophosphorylase. إذا تم تنشيطه بواسطة الأنسولين، فسيستمر سينثاز الجليكوجين في قص الجلوكوز من مركب الجلوكوز UDP إلى بوليمر الجليكوجين.
ميكانيكية التفاعل
يؤثر Phosphoglucomutase أو فسفوجلوكوموتاز على تحول مجموعة فسفوريل عن طريق تبادل مجموعة فوسفوريل مع المادة المتفاعلة.[1] وقد أكدت تجارب وضع العلامات النظائر أن هذا التفاعل يستمر من خلال وسيط الجلوكوز 1,6- ثنائي الفوسفات.[2]
الخطوة الأولى في التفاعل الأمامي هي نقل مجموعة فوسفوريل من الإنزيم إلى جلوكوز 1 فوسفات، وتشكيل جلوكوز 1,6-فوسفات، وترك شكل منزوع الفوسفور من الإنزيم. ثم يخضع الإنزيم لعملية إعادة توجيه انتشارية سريعة لوضع 1-فوسفات البيسفوسفات وسيط بشكل صحيح بالنسبة إلى إنزيم إزالة الفوسفور. أظهرت العلاقات بين الركيزة والسرعة واختبارات النقل المستحث أن الإنزيم المزيل للفوسفور يسهل بعد ذلك نقل مجموعة الفوسفوريل من الجلوكوز 1,6-فوسفات الفوسفات إلى الإنزيم، وتجديد الفسفوجلوكلوتوزات الفوسفاتية وإخراج الجلوكوز 6 في الأمام. ). أكدت الدراسات الهيكلية اللاحقة أن الموقع الوحيد في الإنزيم الذي يتحول إلى فوسفوريلات ومزيل فسفوريلات هو الأكسجين لبقايا سيرين الموقع النشط (انظر الرسم البياني أدناه). مطلوب أيون معدن ثنائي التكافؤ، عادة ما يكون من المغنيسيوم أو الكادميوم، من أجل النشاط الإنزيمي وقد ثبت أنه معقد بشكل مباشر مع مجموعة الفسفوريل التي تم استدراؤها في سيرين الموقع النشط.
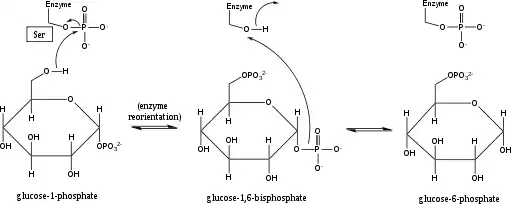
هذا التكوين من وسيط الجلوكوز 1,6- ثنائي الفوسفات مشابه للتحول المتبادل بين 2-فوسفوجليسيرات و 3-فسفوجليسرات محفز بواسطة متغير فوسفوجليسيرات، حيث يتم إنشاء 2،3-ثنائي فوسفوجليسيرات كمركب وسيط.[3]
البناء
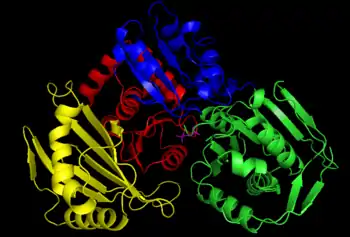
في حين أن فسفوغلوكوموتاز عضلة الأرانب قد خدم كنموذج أولي لكثير من توضيح بنية هذا الإنزيم، تظهر الهياكل البلورية الجديدة المستخلصة من البكتيريا العديد من نفس الخصائص المحددة.[4] يمكن تقسيم كل مونومر فوسفوجلوكوموتاز إلى أربعة مجالات تسلسل، I-IV، بناءً على التكوين المكاني الافتراضي للإنزيم (انظر الصورة على اليمين).[5]
يشتمل كل مونومر على أربع وحدات هيكلية α / distinct مميزة، تحتوي كل منها على واحد من السلاسل الأربعة في كل ورقة من المونومر وتتكون فقط من البقايا في مجال تسلسل معين (انظر الصورة على اليمين).[5] دفن الموقع النشط (بما في ذلك سر-116، وبقايا الحرجة على الانزيم الذي هو فسفرته وdephosphorylated) في مسعور الداخلية من انزيم يعمل على استبعاد المياه من counterproductively hydrolyzing السندات phosphoester حرجة في حين لا يزال يسمح الركيزة الوصول النشطة موقع.[6]
أمراض متعلقة
تحتوي العضلة البشرية على نوعين من الفسفوجلوكوماز مع خصائص تحفيزية متطابقة تقريبًا، PGM I و PGM II.[7] واحد أو آخر من هذه الأشكال مفقود في بعض البشر خلقيًا.[8]
نقص PGM هو حالة نادرة للغاية لا تحتوي على مجموعة من الأعراض الفسيولوجية المميزة. يمكن الكشف عن هذه الحالة من خلال دراسة في المختبر لتحلل السكر اللاهوائي والتي تكشف عن وجود كتلة في المسار نحو إنتاج حمض اللاكتيك بعد الجلوكوز 1-فوسفات ولكن قبل الجلوكوز 6-فوسفات.[9]
يعرف نقص PGM1 باسم متلازمة CDG من النوع 1t (CDG1T، المعروف سابقًا باسم مرض تخزين الجليكوجين من النوع 14 (GSD XIV).[10]
الجينات
- PGM1، PGM2، PGM3، PGM5
انظر أيضًا
المراجع
- Jagannathan, V؛ Luck, JM (1949)، "Phosphoglucomutase; mechanism of action"، Journal of Biological Chemistry، 179 (2): 569–75، PMID 18149991.
- Najjar, V. A.؛ Pullman, M. E. (1954)، "The Occurrence of a Group Transfer Involving Enzyme (phosphoglucomutase) and Substrate"، Science، 119 (3097): 631–4، Bibcode:1954Sci...119..631N، doi:10.1126/science.119.3097.631، PMID 13156640.
- Sutherland, EW؛ Cohn, M (1949)، "The mechanism of the phosphoglucomutase reaction"، Journal of Biological Chemistry، 180 (3): 1285–95، PMID 18148026.
- Mehra-Chaudhary, Ritcha؛ Mick, Jacob؛ Tanner, John J.؛ Henzl, Michael T.؛ Beamer, Lesa J. (2011)، "Crystal structure of a bacterial phosphoglucomutase, an enzyme involved in the virulence of multiple human pathogens"، Proteins: Structure, Function, and Bioinformatics، 79 (4): 1215–29، doi:10.1002/prot.22957، PMID 21246636.
- Dai, JB؛ Liu, Y؛ Ray Jr, WJ؛ Konno, M (1992)، "The crystal structure of muscle phosphoglucomutase refined at 2.7-angstrom resolution"، Journal of Biological Chemistry، 267 (9): 6322–37، PMID 1532581.
- Ray, William J.؛ Puvathingal, Joseph M.؛ Liu, Yiwei (1991)، "Formation of substrate and transition-state analog complexes in crystals of phosphoglucomutase after removing the crystallization salt"، Biochemistry، 30 (28): 6875–85، doi:10.1021/bi00242a011، PMID 1829964.
- Joshi, JG؛ Handler, P (1969)، "Phosphoglucomutase. VI. Purification and properties of phosphoglucomutases from human muscle"، Journal of Biological Chemistry، 244 (12): 3343–51، PMID 4978319.
- Brown DH (1986)، "Glycogen metabolism and glycolysis in muscle"، Myology، New York: McGraw-Hill، ص. 673–95.
- Sugie, H؛ Kobayashi, J؛ Sugie, Y؛ Ichimura, M؛ Miyamoto, R؛ Ito, T؛ Shimizu, K؛ Igarashi, Y (1988)، "Infantile muscle glycogen storage disease: phosphoglucomutase deficiency with decreased muscle and serum carnitine levels"، Neurology، 38 (4): 602–5، doi:10.1212/WNL.38.4.602، PMID 2965317.
- Orphanet: Glycogen storage disease due to phosphoglucomutase deficiency نسخة محفوظة 7 نوفمبر 2017 على موقع واي باك مشين.
روابط خارجية
- Phosphoglucomutase في المكتبة الوطنية الأمريكية للطب نظام فهرسة المواضيع الطبية (MeSH).
 بوابة الكيمياء الحيوية
بوابة الكيمياء الحيوية بوابة علم الأحياء
بوابة علم الأحياء