متلازمة أومين
متلازمة اومين هي جسمية متنحية من نقص المناعة[2] المصاحبة للطفرات الطفولية الضائرة في الجينات ذات الصلة المناعية للخلايا التائية (والخلايا البائية) مثل جينات إعادة التركيب المؤثرة (RAG1 and RAG2), IL-7 Receptor α gene (IL7Rα), DCLRE1C-Artemis, RMRP-CHH, DNA-Ligase IV, common gamma chain, WHN-FOXN1, ZAP-70 and complete DiGeorge anomaly (DiGeorge Syndrome; CHARGE).
| متلازمة أومين | |
|---|---|
 متلازمة أومين لديها نمط متنحي وراثي. متلازمة أومين لديها نمط متنحي وراثي. | |
| معلومات عامة | |
| الاختصاص | علم الدم |
| من أنواع | عوز المناعة المشترك الشديد |
| المظهر السريري | |
| الأعراض | إسهال[1]، وضخامة الكبد والطحال[1] |
الأعراض
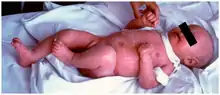
الأعراض هي مشابهة جدا لمرض الطعم ضد المضيف. وذلك لأن المرضى لديهم بعض الخلايا التائية مع مستويات محدودة من إعادة التركيب مع الجينات راج متحولة. هذه الخلايا التائية غير طبيعية ولها تقارب محدد جدا للمستضدات الذاتية الموجودة في الغدة الصعترية وفي المحيط.
هناك أعراض مميزة هي التهاب مزمن في الجلد، والذي يظهر على شكل طفح جلدي أحمر (بداية إريثرودرما). وتشمل الأعراض الأخرى فرط الحمضات والفشل في النمو وتضخم الغدد الليمفاوية وتورم الطحال والإسهال وتضخم الكبد وانخفاض مستويات الغلوبولين المناعي (باستثناء الغلوبولين المناعي E الذي يرتفع)، وانخفاض مستويات الخلايا التائية، وعدم وجود خلايا ب.[3]
علم الوراثة
تسبب متلازمة اومين بفقدان جزئي لوظيفة الجين RAG ويؤدي إلى أعراض مشابهة لمتلازمة نقص المناعة المكتسب الشديد، بما في ذلك العدوى الانتهازية. جينات RAG ضرورية لإعادة التركيب الجيني في مستقبلات الخلايا التائية ومستقبلات الخلايا البائية، وفقدان هذه القدرة يعني أن الجهاز المناعي لديه صعوبة في التعرف على مسببات الأمراض المحددة.[4] وتتميز متلازمة اومين بفقدان وظيفة الخلايا التائية، يمكن أحيانا أن يسبب متلازمة أومن في الجينات إعادة التركيب [5]، بما في ذلك IL-7Rα و RMRP.
العلاج
العلاج الوحيد لمتلازمة اومين هو العلاج الكيميائي تليها زرع نخاع العظم. دون علاج، فإنه قاتل بسرعة في مرحلة الطفولة.
انظر أيضا
- Purine nucleoside phosphorylase deficiency
- قائمة الأمراض والاضطرابات الجلدية
مراجع
- مُعرِّف أنطولوجيا مرض: http://www.disease-ontology.org/?id=DOID:0060010 — تاريخ الاطلاع: 30 نوفمبر 2020 — الرخصة: CC0
- Santagata S, Villa A, Sobacchi C, Cortes P, Vezzoni P (2000)، "The genetic and biochemical basis of Omenn syndrome"، Immunol Rev.، 178: 64–74، doi:10.1034/j.1600-065X.2000.17818.x، PMID 11213808.
- Geha, Raif؛ Notarangelo, Luigi (2012)، Case Studies in Immunology: A Clinical Companion (ط. 6th)، Garland Science، ISBN 978-0-8153-4441-4.
- Parham, Peter (2009)، The Immune System (ط. 3rd)، Taylor & Francis Group، ص. 128، ISBN 9781136977107.
- Lev A, Simon AJ, Ben-Ari J, Takagi D, Stauber T, Trakhtenbrot L, Rosenthal E, Rechavi G, Amariglio N, Somech R (2014)، "Co-existence of clonal expanded autologous and transplacental-acquired maternal T cells in recombination activating gene-deficient severe combined immunodeficiency."، Clin Exp Immunol.، 176 (3): 380–6، doi:10.1111/cei.12273، PMC 4008982، PMID 24666246.
 بوابة طب
بوابة طب