متلازمة بركات
يعتبر مرض متلازمة بركات مرضاً نادراً يتميز بكسل الغدة جار الدرقية والصمم الحسي العصبي وأمراض الكلى، و يعرف أيضًا باسم متلازمة HDR[1] حيث تم وصفه لأول مرة من قبل أمين بركات في عام 1977.[2]
| متلازمة بركات | |
|---|---|
 متلازمة بركات | |
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية |
| من أنواع | تشوه متلازمي كلوي أو في السبيل البولي |
| التاريخ | |
| سُمي باسم | أمين بركات |
وصف المرض
تعتبر متلازمة بركات اضطراب[3] تطوري وراثي مع تنوع سريري يتميز بكسل الغدة جار الدرقية والصمم الحسي العصبي وأمراض الكلى،[4] حيث يظهر على الأشخاص المصابون بها عادة نقص كالسيوم الدم أو التكزز أو تشنجات من غيرالحمى في أي عمر، وعادةً ما يكون ضعف السمع ثنائيًا وقد يتراوح من ضعف خفيف إلى عميق. وبالمقابل تشمل أمراض الكلى :المتلازمة الكلوية، وتكيس الكلى، وخلل التنسج الكلوي، ونقص تنسج أو عدم تنسج الحوض وتشوهه، والارتجاع المثاني الحالبي، وأمراض الكلى المزمنة، والبيلة الدموية، وزلال البول، والتندب الكلوي.
تشمل الأعراض الأخرى المسجلة: الإعاقة الذهنية، وتكيس المبايض، والخصائص المميزة للوجه، والسكتة الدماغية، والتهاب الشبكية الصباغي.
علم الوراثة
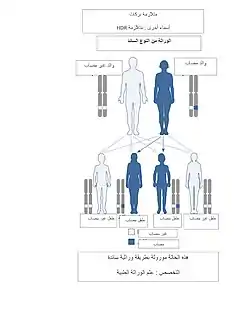
تم نسب الخلل في معظم الحالات إلى الكروموسوم 10p (موقع خريطة الجينات: 10pter-p13 أو 10p14-p15.1)، و يبدو أن عدم الكفاية (حذف) لعامل نسخ إصبع الزنك (جاتا٣) أو الطفرات في جين (جاتا٣) [5]هو السبب الكامن وراء هذه المتلازمة، حيث يسبب فشل في تحديد المجال الحسي ويؤدي بالتالي إلى زيادة موت الخلايا في قناة القوقعة وبالتالي يؤدي إلى الصمم،[6] ونظرًا لأن طيف التباين الظاهري في الأشخاص المصابين كبير جدًا، فمن المحتمل أن تنشأ متلازمة بركات (HDR) كإضطراب منخفض الإنتشار لنقص عامل نسخ اصبع الزنك حيث تلعب خلفية هؤلاء الأشخاص الجينية دورًا رئيسيًا في شدة المرض ومن المحتمل أن تكون الوراثة من النوع السائد.
التشخيص
يجب إجراء تشخيص شامل لكل فرد مصاب، ويجب فحص اشقاء المصابين للتأكد من عدم الإصابة بالصمم وأمراض الغدة الدرقية أو أمراض الكلى ويجب ايضاً أخذ المتلازمة في الإعتبار عند فحص الرضع الذين تم تشخيصهم قبل الولادة بعيب كروموسوم 10p، وأولئك الذين تم تشخيصهم بأعراض واضحة ومحددة لتشوهات المسالك البولية.
العلاج
تتكون خطة العلاج من علاج التشوهات الظاهرة في وقت الفحص حيث تشمل الإستشارة الوراثية وتصحيح الكالسيوم وعلاج مشاكل السمع ومراقبة وظائف الكلى والمراقبة الدقيقة لتكيسات الكلى.
توقعات سير المرض
يعتمد التوقع على شدة مرض الكلى، ولا يتأثر متوسط العمر المتوقع إذا كانت شدة المرض خفيفة.
علم الاوبئة
معدل إنتشار المرض غير معروف، ولكن يعتبر المرض نادرًا جدًا.[7] وقد لوحظ إصابة كل من الذكور والإناث به، عن طريق ظهور أعراض كسل الغدة جار الدرقية، والصمم الحسي العصبي، وأمراض الكلى عليهم، ومع ذلك، يمكن أن تختلف الأعراض المحددة والشدة حيث يعاني حوالي 65٪ من المصابين بمتلازمة بركات من كسل الغدة جار الدرقية والصمم الحسي العصبي وأمراض الكلى معًا.
المراجع
- "Hypoparathyroidism sensorineural deafness renal disease syndrome". www.orpha.net. Retrieved 29 March 2019.
- Barakat AY, D'Albora JB, Martin MM, Jose PA (July 1977). "Familial nephrosis, nerve deafness, and hypoparathyroidism". J. Pediatr. 91 (1): 61–4. doi:10.1016/S0022-3476(77)80445-9. PMID 874665.
- William B. Coleman (9 February 2010). Essential Concepts in Molecular Pathology. Academic Press. pp. 297–. ISBN 978-0-12-374418-0. Retrieved 5 December 2010.
- "Barakat syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. National Institutes of Health. 7 September 2018. Retrieved 12 November 2019.
- Bernardini L, Sinibaldi L, Capalbo A, et al. (July 2009). "HDR (Hypoparathyroidism, Deafness, Renal dysplasia) syndrome associated to GATA3 gene duplication". Clin. Genet. 76 (1): 117–9. doi:10.1111/j.1399-0004.2009.01170.x. PMID 19659764.
- Luo, X.-j.; Deng, M.; Xie, X.; Huang, L.; Wang, H.; Jiang, L.; Liang, G.; Hu, F.; Tieu, R.; Chen, R.; Gan, L. (10 May 2013). "GATA3 controls the specification of prosensory domain and neuronal survival in the mouse cochlea". Human Molecular Genetics. 22 (18): 3609–3623. doi:10.1093/hmg/ddt212. PMC 3749857. PMID 23666531.
- Ranjbar-Omrani, G; Zamiri, N; Sabayan, B; Mohammadzadeh, A (May 2008). "Concomitant hypoparathyroidism, sensorineural deafness, and renal agenesis: a case of Barakat syndrome". Archives of Iranian Medicine. 11 (3): 337–40. PMID 18426329.
 بوابة طب
بوابة طب