Pi helix
A pi helix (or π-helix) is a type of secondary structure found in proteins. Discovered by crystallographer Barbara Low in 1952[1] and once thought to be rare, short π-helices are found in 15% of known protein structures and are believed to be an evolutionary adaptation derived by the insertion of a single amino acid into an α-helix.[2] Because such insertions are highly destabilizing,[3] the formation of π-helices would tend to be selected against unless it provided some functional advantage to the protein. π-helices therefore are typically found near functional sites of proteins.[2][4][5][6]
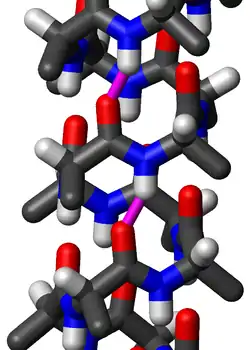
Standard structure
The amino acids in a standard π-helix are arranged in a right-handed helical structure. Each amino acid corresponds to an 87° turn in the helix (i.e., the helix has 4.1 residues per turn), and a translation of 1.15 Å (0.115 nm) along the helical axis. Most importantly, the N-H group of an amino acid forms a hydrogen bond with the C=O group of the amino acid five residues earlier; this repeated i + 5 → i hydrogen bonding defines a π-helix. Similar structures include the 310 helix (i + 3 → i hydrogen bonding) and the α-helix (i + 4 → i hydrogen bonding).

The majority of π-helices are only 7 residues in length and do not adopt regularly repeating (φ, ψ) dihedral angles throughout the entire structure like that of α-helices or β-sheets. Because of this, textbooks that provide single dihedral values for all residues in the π-helix are misleading. Some generalizations can be made, however. When the first and last residue pairs are excluded, dihedral angles exist such that the ψ dihedral angle of one residue and the φ dihedral angle of the next residue sum to roughly −125°. The first and last residue pairs sum to −95° and −105°, respectively. For comparison, the sum of the dihedral angles for a 310 helix is roughly −75°, whereas that for the α-helix is roughly −105°. Proline is often seen immediately following the end of π-helices. The general formula for the rotation angle Ω per residue of any polypeptide helix with trans isomers is given by the equation

Left-handed structure
In principle, a left-handed version of the π-helix is possible by reversing the sign of the (φ, ψ) dihedral angles to (55°, 70°). This pseudo-"mirror-image" helix has roughly the same number of residues per turn (4.1) and helical pitch (1.5 Å [150 pm]). It is not a true mirror image, because the amino-acid residues still have a left-handed chirality. A long left-handed π-helix is unlikely to be observed in proteins because, among the naturally occurring amino acids, only glycine is likely to adopt positive φ dihedral angles such as 55°.
π-helices in nature
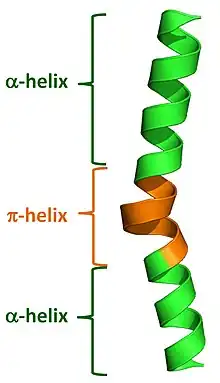
Commonly used automated secondary structure assignment programs, such as DSSP, suggest <1% of proteins contain a π-helix. This mis-characterization results from the fact that naturally occurring π-helices are typically short in length (7 to 10 residues) and are almost always associated with (i.e. flanked by) α-helices on either end. Nearly all π-helices are therefore cryptic in that the π-helical residues are incorrectly assigned as either α-helical or as "turns". Recently developed programs have been written to properly annotate π-helices in protein structures and they have found that 1 in 6 proteins (around 15%) do in fact contain at least one π-helical segment.[2]
Natural π-helices can easily be identified in a structure as a "bulge" within a longer α-helix. Such helical bulges have previously been referred to as α-aneurisms, α-bulges, π-bulges, wide-turns, looping outs and π-turns, but in fact are π-helices as determined by their repeating i + 5 → i hydrogen bonds.[2] Evidence suggests that these bulges, or π-helices, are created by the insertion of a single additional amino acid into a pre-existing α-helix. Thus, α-helices and π-helices can be inter-converted by the insertion and deletion of a single amino acid.[2][3] Given both the relatively high rate of occurrence of π-helices and their noted association with functional sites (i.e. active sites) of proteins, this ability to interconvert between α-helices and π-helices has been an important mechanism of altering and diversifying protein functionality over the course of evolution.[2]
One of the most notable group of proteins whose functional diversification appears to have been heavily influenced by such an evolutionary mechanism is the ferritin-like superfamily, which includes ferritins, bacterioferritins, rubrerythrins, class I ribonucleotide reductases and soluble methane monooxygenases. Soluble methane monooxygenase is the current record holder for the most number of π-helices in a single enzyme with 13 (PDB code 1MTY). However, the bacterial homologue of a Na+/Cl− dependent neurotransmitter transporter (PDB code 2A65) holds the record for the most π-helices in a single peptide chain with 8.[2]
See also
References
- "(IUCr) Barbara Wharton Low (1920-2019)". www.iucr.org. Retrieved 2019-10-02.
- Cooley RB, Arp DJ, Karplus PA (2010). "Evolutionary origin of a secondary structure: π-helices as cryptic but widespread insertional variations of α-helices enhancing protein functionality". J Mol Biol. 404 (2): 232–246. doi:10.1016/j.jmb.2010.09.034. PMC 2981643. PMID 20888342.
- Keefe LJ, Sondek J, Shortle D, Lattman EE (2000). "The alpha aneurism: a structural motif revealed in an insertion mutant of staphylococcal nuclease". Proc. Natl. Acad. Sci. U.S.A. 90 (8): 3275–3279. doi:10.1073/pnas.90.8.3275. PMC 46282. PMID 8475069.
- Weaver TM (2000). "The pi-helix translates structure into function". Protein Science. 9 (1): 201–206. doi:10.1110/ps.9.1.201. PMC 2144447. PMID 10739264.
- Fodje MN, Al-Karadaghi S (2002). "Occurrence, conformational features and amino acid propensities for the pi-helix". Protein Eng. 15 (5): 353–358. doi:10.1093/protein/15.5.353. PMID 12034854.
- Mohapatra, Samar Bhallabha; Manoj, Narayanan (January 2021). "A conserved π-helix plays a key role in thermoadaptation of catalysis in the glycoside hydrolase family 4". Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 1869 (1): 140523. doi:10.1016/j.bbapap.2020.140523. PMID 32853774. S2CID 221358131.
| Curves |  | ||
|---|---|---|---|
| Helices |
| ||
| Spirals | |||