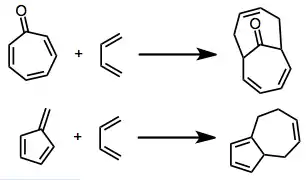
6+4 Cycloaddition
[6+4] Cycloaddition is a type of cycloaddition between a six-atom pi system and a four-atom pi system, leading to a ten-membered ring. Because this is a higher-order cycloaddition, issues of periselectivity arise in addition to the usual concerns about regio- and stereoselectivity. Six-atom pi systems that have been employed in the reaction include tropone and tropone derivatives, fulvenes, and cycloheptatriene cobalt complexes.[1]
Introduction
[6+4] Cycloaddition is a thermally allowed, higher-order cycloaddition process leading to ten-membered rings. Although most linear, acyclic trienes do not give [6+4] products selectively, cyclic trienes give high yields of [6+4] products in many cases. Both cycloheptatrienes and fulvenes can be employed in this reaction, and electron-deficient tropones in particular work well. The pericyclic and transition-metal-mediated versions of the reaction are stereocomplementary: the former gives exo products, and the latter endo products, with essentially complete selectivity in nearly all cases. The possibility of building complex carbocyclic frameworks efficiently has made this reaction particularly attractive synthetically.[2]
(1)
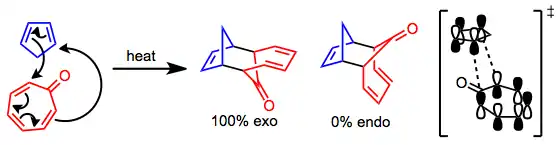
Mechanism and stereochemistry
Metal-free reaction
The metal-free version of the [6+4] cycloaddition takes place through a concerted, pericyclic process. The frontier molecular orbitals involved in reactions of tropone illustrate that a repulsive secondary orbital interaction likely destabilizes the endo transition state, leading to complete selectivity for exo products.[3]
(2)
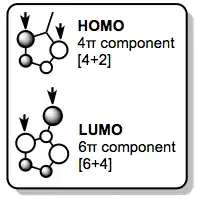
Fulvenes react similarly, although selective [6+4] reactions require the use of an electron-rich diene. Fulvene's frontier orbitals illustrate that it will only act as a 6π component in LUMO-controlled reactions. The next-highest occupied molecular orbital (NHOMO, not shown) also has the proper symmetry and orbital coefficients to participate in [6+4] cycloaddition; the NHOMO can be activated for reaction by substituting the exo methylene with electron-donating groups.[4]
(3)
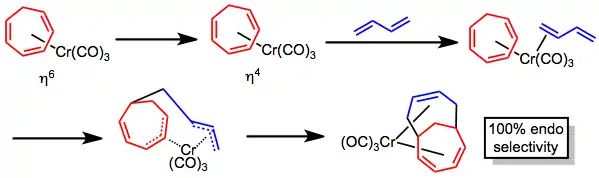
Metal-promoted reaction
The metal-promoted [6+4] cycloaddition using a cycloheptatriene–metal complex is a stepwise, photolytic process that lacks the periselectivity issues of the purely thermal [6+4] cycloaddition. The mechanism has been debated, but likely proceeds through a hapticity change in the metal-triene complex followed by coordination of the diene component and coupling. Dissociation of carbon monoxide has also been invoked instead of hapticity change, but the evolution of carbon monoxide is not observed during the reaction. Complete endo selectivity is observed for this process as a result of templating by the metal.[5]
(4)
Although notable work on [6+4] cycloadditions was reported by Rigby et al. in the late 1980s, the study of metal-complexed dienes with cycloheptatriene received comparatively little attention. In 2006, Welker and co-workers reported that their cobaloxime dienes react with unsubstituted tropones through exo transition states exclusively. The same dienes were shown to react in both [6+4] and [4+2] cycloadditions when the reacting tropone has at least one electron withdrawing group (EWG). The [4+2] reaction pathway is dominant when the tropone has substituents at the bond forming centers and EWGs.[6]
Scope and limitations
[6+4] Cycloaddition may be carried out using tropones, fulvenes, or chromium complexes of cycloheptatrienes. The scope of 4π reaction partners is broad, but limited in some cases by the electronic bias of the 6π component. For instance, cycloadditions of tropones are generally higher yielding when an electron-rich diene is involved.[7] Electron-rich fulvenes react well with electron-poor dienes, and vice versa.[8] Yields of transition-metal-mediated cycloadditions show a relatively weak dependence on the electronic nature of the substrates[9] and yields are generally high.
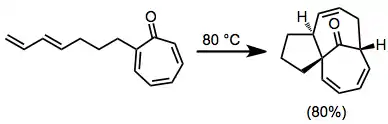
Substituted tropones can be problematic substrates for [6+4] cycloadditions. One method addressing this problem is the intramolecular [6+4], which employs tethered dienes. In addition, this method has the potential to generate complex, polycyclic frameworks.[10]
(5)
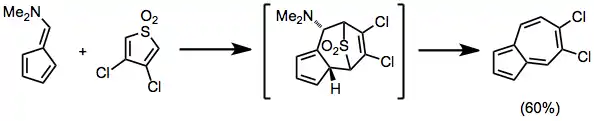
Unsubstituted fulvenes usually only react with electron-rich dienes in the [6+4] mode; however, substituting the fulvene with an electron-donating group in the 6 position facilitates reaction with electron-poor dienes via the fulvene NHOMO. Cheletropic extrusion of sulfur dioxide occurs under the reaction conditions below.[11]
(6)
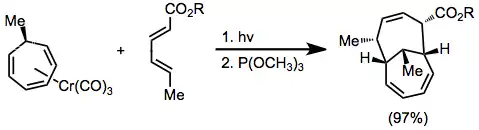
Existing stereochemistry in the diene or triene can be used for diastereoselective transformations that set up to four stereocenters in a single step.[12]
(7)
Comparison with other methods
Most methods that form the ring systems accessible through [6+4] cycloaddition (bicyclo[4.4.1]undecanes and bicyclo[5.3.0]decanes) require multiple steps, harsh conditions, or extensive synthetic manipulations. These methods also may lack the stereoselectivity of the [6+4] process. The methods shown below are representative of alternate strategies and highlight some of the difficulties associated with them.[13][14]
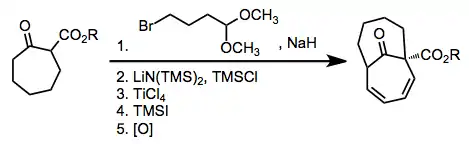
(8)
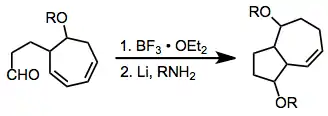
(9)
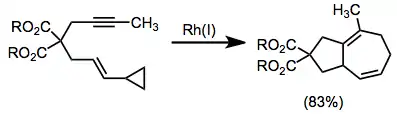
Rhodium-catalyzed annulation of alkynes with vinylcyclopropanes provides the bicyclo[5.3.0]decane with a unique saturation pattern.[15]
(10)
Experimental conditions and procedure
Typical conditions
Thermal [6+4] reactions are typically carried out at reflux in a non-polar solvent such as xylene, benzene, or toluene. As higher temperatures may promote lower-order processes, the temperature should be kept as low as possible. Metal-mediated reactions are usually carried out in hexanes, with a small amount of ether for solubility purposes if necessary. Pyrex and uranium glass-filtered light give higher yields than quartz-filtered light.
See also
References
- Rigby, J. H. Org. React. 1997, 49, 331. doi:10.1002/0471264180.or049.02
- Rigby, J. H. in Comprehensive Organic Synthesis, Trost, B. M.; Fleming, I., Eds., Vol. 5, Pergamon, Oxford, 1991, pp. 617–643.
- Mukherjee, D.; Watts, C. R.; Houk, K. N. J. Org. Chem. 1978, 43, 817.
- Houk, K. N.; Sims, J.; Watts, C. R.; Luskus, L. J. J. Am. Chem. Soc. 1973, 95, 7301.
- Michels, E.; Sheldrick, W.S.; Kreiter, C.G. Chem. Ber. 1985, 118, 964.
- Pidaparthi, R. R.; Welker, M. E.; Day, C. S. Organometallics 2006, 25, 974.
- Garst, M. E.; Roberts, V. A.; Prussin, C. Tetrahedron 1983, 39, 581.
- Wu, T. -C.; Mareda, J.; Gupta, Y. N.; Houk, K. N. J. Am. Chem. Soc. 1983, 105, 6996.
- Ref. 1, pp. 344.
- Funk, R. L.; Bolton, G. L. J. Am. Chem. Soc. 1986, 108, 4655.
- Reiter, S. E.; Dunn, L. C.; Houk, K. N. J. Am. Chem. Soc. 1977, 99, 4199.
- Rigby, J. H.; Ateeq, H. S.; Charles, N. R.; Cuisiat, S. V.; Ferguson, M. D.; Henshilwood, J. A.; Krueger, A. C.; Ogbu, C. O.; Short, K. M.; Heeg, M. J. J. Am. Chem. Soc. 1993, 115, 1382.
- Paquette, L. A.; Nitz, T. J.; Ross, R. J.; Springer, J. P. J. Am. Chem. Soc. 1984, 106, 1446.
- Rigby, J. H.; Wilson, J. Z. J. Am. Chem. Soc. 1984, 106, 8217.
- Wender, P. A.; Takahashi, H.; Witulski, B. J. Am. Chem. Soc. 1995, 117, 4720.