Alpha-aminoadipic semialdehyde synthase
Alpha-aminoadipic semialdehyde synthase is an enzyme encoded by the AASS gene in humans and is involved in their major lysine degradation pathway. It is similar to the separate enzymes coded for by the LYS1 and LYS9 genes in yeast, and related to, although not similar in structure, the bifunctional enzyme found in plants.[5][6] In humans, mutations in the AASS gene, and the corresponding alpha-aminoadipic semialdehyde synthase enzyme are associated with familial hyperlysinemia.[5][7][8] This condition is inherited in an autosomal recessive pattern and is not considered a particularly negative condition, thus making it a rare disease.[9]
| AASS | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | AASS, LKR/SDH, LKRSDH, LORSDH, aminoadipate-semialdehyde synthase | ||||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | OMIM: 605113 MGI: 1353573 HomoloGene: 4212 GeneCards: AASS | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||



Function
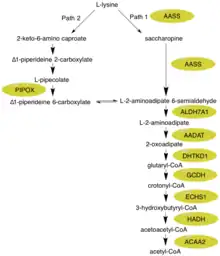
The alpha-aminoadipic semialdehyde synthase protein catalyzes the first two steps in the mammalian L-lysine degradation via saccharopine pathway within the mitochondria, which is thought to be the main metabolic route for lysine degradation in upper eukaryotes.[11][12] The specific subpathway that this enzyme focuses on is the synthesis of glutaryl-CoA from L-lysine.[9] Glutaryl-CoA can act as an intermediate in a more expanded conversion/degradation pathway from L-lysine to acetyl-CoA.
Two noticeable components of the L-lysine degradation via saccharopine pathway are the intermediately-used reaction/product glutamate and the eventual carbon sink acetyl-CoA. Glutamate is an important compound within the body which acts as a neurotransmitter tied to learning and Huntington's disease.[13][14] Acetyl-CoA is arguably of an even higher level of importance, acting as one of the integral components of the Citric Acid/Kreb cycle, with the primary function of delivering an acetyl group to be oxidized for energy production.[15] Thus, the function of alpha-aminoadipic semialdehyde synthase is tied to the levels of two integral compounds within the body.
Mechanism
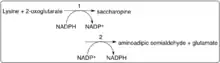
First, the N-terminal portion of this enzyme which contains lysine-ketoglutarate reductase (LOR/LKR) activity (EC:1.5.1.8) condenses lysine and 2-oxoglutarate to a molecule called saccharopine (Reaction 1 on the figure to the right).[7][11] Then, the C-terminal portion of this enzyme, which contains saccharopine dehydrogenase (SHD) activity (EC:1.5.1.9), catalyzes the oxidation of saccharopine to produce alpha-aminoadipic semialdehyde and glutamate (Reaction 2 on the figure to the right).[7][11] Note: These reactions are the reverse of the corresponding steps in the lysine biosynthesis pathways present in yeast and fungi.[16][17][18]
These reactions can be visualized as well in reaction equation form:
N(6)-(L-1,3-dicarboxypropyl)-L-lysine + NADP+ + H2O = L-lysine + 2-oxoglutarate + NADPH followed by
N(6)-(L-1,3-dicarboxypropyl)-L-lysine + NAD+ + H2O = L-glutamate + (S)-2-amino-6-oxohexanoate + NADH.[9]
Structure
The native human enzyme is bifunctional, much like the LKR/SHD found in plants, and thus, is thought to be similar in structure.[16] The bifunctionality of this enzyme comes from the fact that it contains two distinct active sites, one at its C-terminal, and one at its N-terminal.[7] The C-terminal portion of alpha-aminoadipic semialdehyde synthase contains the SHD activity and the N-terminal portion contains LKR.[19] To date, a structure of alpha-aminoadipic semialdehyde synthase has not been determined.[20] The enzyme does not have linker region present in plants between its C and N-termini, so theories suggest the actual structure contains an LKR-activity region bound to an SHD-activity region, like that in Magnaporthe grisea.[19]
Disease relevance
Alpha-aminoadipic semialdehyde synthase is encoded for by the AASS gene, and mutations in this gene lead to hyperlysinemia.[5][7] This is characterized by impaired breakdown of lysine which results in elevated levels of lysine in the blood and urine. These increased levels of lysine do not appear to have any negative effects on the body.[8] Other names for this condition include:[8]
- alpha-aminoadipic semialdehyde deficiency disease
- familial hyperlysinemia
- lysine alpha-ketoglutarate reductase deficiency disease
- saccharopine dehydrogenase deficiency disease
- saccharopinuria
Hyperlysinemia is characterized by elevated plasma lysine levels that exceed 600 μmol/L and can reach up to 2000 μmol/L.[21][22] These increased levels of lysine do not appear to have any negative effects on the body.[8] The main reason for this is that several alternative biochemical reactions can take place. First, lysine can be used in place of ornithine in the urea cycle resulting in the production of homoarginine.[23] Additionally, even though most mammals use the saccharopine pathway for most lysine degradation (Path 1), the brain has an alternative pathway (Path 2) which goes through an L-pipecolic acid intermediate - both of these can be seen in the figure.[23] It is important to note that Path 1 takes place in the mitochondria while Path 2 takes places in the peroxisome.[12] Looking at other key enzymes within the L-lysine degradation pathway, ALDH7A1 is deficient in children with pyridoxine-dependent seizures.[24] GCDH is deficient in glutaric aciduria type 1.[25] The intermediate 2-oxoadipate is metabolized by 2-oxoadipate dehydrogenase, resembling the Citric Acid/Kreb cycle enzyme complex 2-oxoglutarate dehydrogenase.[10]
Two types of familial hyperlysinemia have been described so far: type I is associated with a combined deficiency of the two enzyme activities, LOR and SDH, whereas in familial hyperlysinemia type II only the saccharopine dehydrogenase activity is impaired.[26][27] Type II hyperlysinemia is also referred to as saccharopinuria.[10]
An additional condition shown to be related to hyperlysinemia is dienoyl-CoA reductase deficiency, though this is a relatively recent discovery and there are not many publications supporting this.[28]
References
- GRCh38: Ensembl release 89: ENSG00000008311 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000029695 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Sacksteder KA, Biery BJ, Morrell JC, Goodman BK, Geisbrecht BV, Cox RP, Gould SJ, Geraghty MT (June 2000). "Identification of the alpha-aminoadipic semialdehyde synthase gene, which is defective in familial hyperlysinemia". American Journal of Human Genetics. 66 (6): 1736–43. doi:10.1086/302919. PMC 1378037. PMID 10775527.
- Zhu X, Tang G, Galili G (December 2002). "The activity of the Arabidopsis bifunctional lysine-ketoglutarate reductase/saccharopine dehydrogenase enzyme of lysine catabolism is regulated by functional interaction between its two enzyme domains". The Journal of Biological Chemistry. 277 (51): 49655–61. doi:10.1074/jbc.M205466200. PMID 12393892.
- "Entrez Gene: AASS aminoadipate-semialdehyde synthase".
- "hyperlysinemia". Genetics Home Reference. Retrieved 2017-03-04.
- "Alpha-aminoadipic semialdehyde synthase, mitochondrial". UniProt. Retrieved 2017-03-04.
- Houten SM, Te Brinke H, Denis S, Ruiter JP, Knegt AC, de Klerk JB, Augoustides-Savvopoulou P, Häberle J, Baumgartner MR, Coşkun T, Zschocke J, Sass JO, Poll-The BT, Wanders RJ, Duran M (April 2013). "Genetic basis of hyperlysinemia". Orphanet Journal of Rare Diseases. 8: 57. doi:10.1186/1750-1172-8-57. PMC 3626681. PMID 23570448.
- Papes F, Kemper EL, Cord-Neto G, Langone F, Arruda P (December 1999). "Lysine degradation through the saccharopine pathway in mammals: involvement of both bifunctional and monofunctional lysine-degrading enzymes in mouse". The Biochemical Journal. 344 (2): 555–63. doi:10.1042/0264-6021:3440555. PMC 1220675. PMID 10567240.
- Danhauser K, Sauer SW, Haack TB, Wieland T, Staufner C, Graf E, Zschocke J, Strom TM, Traub T, Okun JG, Meitinger T, Hoffmann GF, Prokisch H, Kölker S (December 2012). "DHTKD1 mutations cause 2-aminoadipic and 2-oxoadipic aciduria". American Journal of Human Genetics. 91 (6): 1082–7. doi:10.1016/j.ajhg.2012.10.006. PMC 3516599. PMID 23141293.
- Meldrum BS (April 2000). "Glutamate as a neurotransmitter in the brain: review of physiology and pathology". The Journal of Nutrition. 130 (4S Suppl): 1007S–15S. doi:10.1093/jn/130.4.1007s. PMID 10736372. Archived from the original on 2017-05-16. Retrieved 2017-03-05.
- "About Glutamate Toxicity". Huniting Disease Outreach for Education at Stanford (HOPES). Huntington’s Disease Society of America. 26 June 2011. Retrieved 2017-03-05.
- Ophardt CE (2003). "Acetyl CoA Crossroads Compound". Virtual ChemBook. Elmhurst College. Archived from the original on 2016-11-15. Retrieved 2017-03-05.
- Markovitz PJ, Chuang DT, Cox RP (October 1984). "Familial hyperlysinemias. Purification and characterization of the bifunctional aminoadipic semialdehyde synthase with lysine-ketoglutarate reductase and saccharopine dehydrogenase activities". The Journal of Biological Chemistry. 259 (19): 11643–6. doi:10.1016/S0021-9258(20)71252-4. PMID 6434529.
- Jones EE, Broquist HP (June 1965). "Saccharopine, an intermediate of the aminoadipic acid pathway of lysine biosynthesis. Ii. studies in Saccharomyces cereviseae [sic]". The Journal of Biological Chemistry. 240 (6): 2531–6. doi:10.1016/S0021-9258(18)97358-8. PMID 14304864.
- Trupin JS, Broquist HP (June 1965). "Saccharopine, an intermediate of the aminoadipic acid pathway of lysine biosynthesis. I. studies in neurospora crassa". The Journal of Biological Chemistry. 240 (6): 2524–30. doi:10.1016/S0021-9258(18)97357-6. PMID 14304863.
- Johansson E, Steffens JJ, Lindqvist Y, Schneider G (October 2000). "Crystal structure of saccharopine reductase from Magnaporthe grisea, an enzyme of the alpha-aminoadipate pathway of lysine biosynthesis". Structure. 8 (10): 1037–47. doi:10.1016/s0969-2126(00)00512-8. PMID 11080625.
- "AASS - aminoadipate-semialdehyde synthase". RCSB Protein Data Bank. Archived from the original on 2017-11-07. Retrieved 2017-03-06 – via PDB.
- Hoffmann GF, Kolker S (2012). "Cerebral organic acid disorders and other disorders of lysine catabolism". In Saudubray JM, van den Berghe G, Walter JH (eds.). Inborn metabolic diseases diagnosis and treatment (5th ed.). Berlin: Springer. pp. 333–346. ISBN 978-3-642-15720-2.
- Saudubray JM, Rabier D (June 2007). "Biomarkers identified in inborn errors for lysine, arginine, and ornithine". The Journal of Nutrition. 137 (6 Suppl 2): 1669S–1672S. doi:10.1093/jn/137.6.1669S. PMID 17513445.
- vd Heiden C, Brink M, de Bree PK, v Sprang FJ, Wadman SK, de Pater JM, van Biervliet JP (1978). "Familial hyperlysinaemia due to L-lysine alpha-ketoglutarate reductase deficiency: results of attempted treatment". Journal of Inherited Metabolic Disease. 1 (3): 89–94. doi:10.1007/bf01805679. PMID 116084. S2CID 35326745.
- Mills PB, Struys E, Jakobs C, Plecko B, Baxter P, Baumgartner M, Willemsen MA, Omran H, Tacke U, Uhlenberg B, Weschke B, Clayton PT (March 2006). "Mutations in antiquitin in individuals with pyridoxine-dependent seizures". Nature Medicine. 12 (3): 307–9. doi:10.1038/nm1366. PMID 16491085. S2CID 27940375.
- Goodman SI, Kratz LE, DiGiulio KA, Biery BJ, Goodman KE, Isaya G, Frerman FE (September 1995). "Cloning of glutaryl-CoA dehydrogenase cDNA, and expression of wild type and mutant enzymes in Escherichia coli". Human Molecular Genetics. 4 (9): 1493–8. doi:10.1093/hmg/4.9.1493. PMID 8541831.
- Dancis J, Hutzler J, Cox RP (May 1979). "Familial hyperlysinemia: enzyme studies, diagnostic methods, comments on terminology". American Journal of Human Genetics. 31 (3): 290–9. PMC 1685795. PMID 463877.
- Cederbaum SD, Shaw KN, Dancis J, Hutzler J, Blaskovics JC (August 1979). "Hyperlysinemia with saccharopinuria due to combined lysine-ketoglutarate reductase and saccharopine dehydrogenase deficiencies presenting as cystinuria". The Journal of Pediatrics. 95 (2): 234–8. doi:10.1016/s0022-3476(79)80657-5. PMID 571908.
- Houten SM, Denis S, Te Brinke H, Jongejan A, van Kampen AH, Bradley EJ, Baas F, Hennekam RC, Millington DS, Young SP, Frazier DM, Gucsavas-Calikoglu M, Wanders RJ (September 2014). "Mitochondrial NADP(H) deficiency due to a mutation in NADK2 causes dienoyl-CoA reductase deficiency with hyperlysinemia". Human Molecular Genetics. 23 (18): 5009–16. doi:10.1093/hmg/ddu218. PMID 24847004.
Further reading
- Maruyama K, Sugano S (January 1994). "Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides". Gene. 138 (1–2): 171–4. doi:10.1016/0378-1119(94)90802-8. PMID 8125298.
External links
- AASS human gene location in the UCSC Genome Browser.
- AASS human gene details in the UCSC Genome Browser.