Axenfeld–Rieger syndrome
Axenfeld–Rieger syndrome is a rare autosomal dominant[2] disorder, which affects the development of the teeth, eyes, and abdominal region.[3]
| Axenfeld–Rieger syndrome | |
|---|---|
| Other names | Axenfeld syndrome, Hagedoom syndrome |
 | |
| a) Microdontia and hypodontia. b) Slit pupil and iris atrophy right eye. c) Corectopia with iris atrophy left eye. d) Posterior embryotoxon right eye. e) Posterior embryotoxon left eye. f) Broad peripheral anterior synechiae right eye.[1] | |
| Specialty | Medical genetics |
Axenfeld–Rieger syndrome is part of the so-called iridocorneal or anterior segment dysgenesis syndromes,[4] which were formerly known as anterior segment cleavage syndromes, anterior chamber segmentation syndromes or mesodermal dysgenesis. Although the exact classification of this set of signs and symptoms is somewhat confusing in current scientific literature, most authors agree with the classification cited here. Axenfeld Anomaly is known as the development of a posterior embryotoxon, associated with strands of the iris adhered to a Schwalbe line that has been displaced anteriorly,[5] which when added to glaucoma is called Axenfeld Syndrome. Rieger's Anomaly is defined by a universe of congenital anomalies of the iris, such as iris hypoplasia, corectopia or polycoria.[6] When systemic findings are added to Rieger's anomaly, such as bone, facial and/or dental defects, it is known as Rieger syndrome. The combination of both entities gives rise to the Axenfeld-Rieger Anomaly when there are no systemic abnormalities and Axenfeld-Rieger Syndrome when there are.[7]
Axenfeld-Rieger Syndrome, is a rare disease that affects the eye bilaterally, with an estimated prevalence of 1/200,000 people, without gender predilection, and is characterized by autosomal dominant inheritance with complete penetrance of variable expressivity. The genes that have been identified in approximately 50% of cases are PITX2 and FOXC1.[8][9][10] Given the important hereditary factor, it is important to evaluate the most direct members of the family.
To explain the ocular alterations, there is a theory of the mechanism postulated by Shields et al.,[7] which implies an arrest in the migration of neural crest cells towards the third trimester of gestation, which leads to the persistence of primordial endothelial tissue in the iris and anterior chamber angle. Contraction of these membranes after birth lead to the progressive changes seen in some patients. This primordial endothelium also generates an excessive and atypical basement membrane, especially near the limbal corneal junction, which accounts for the prominent Schwalbe line. In the case of secondary glaucoma, it would be the consequence of dysgenesis in the chamber sinus.
Signs and symptoms
Disease manifestations:
Regarding the age of diagnosis, this differs according to the intensity of the symptoms, ranging from asymptomatic to florid symptoms, characterized by ocular and systemic diseases, affecting multiple organs that have in common their origin in the neural crest.
Eye manifestations: Bilateral ocular manifestations are usually pathognomonic of the disease. In the case of children who develop glaucoma,[11] they may attend the consultation with signs and symptoms of buphthalmos, photosensitivity, tearing, corneal decompensation, which associated with poor vision, can be completed with a strabismus.[12] In the case of the adult, there is a greater chance of not presenting symptoms, so an ophthalmological control may be required to detect the problem. Using a slit lamp, a posterior embryotoxon characterized by a prominent anteriorly displaced Schwalbe's ring near the temporal corneal limbus can be revealed.[5] The unexpected finding of a posterior embryotoxon as a single whitish irregular arcuate ridge, on routine examination, is not necessarily a diagnosis of ARS, as this occurs in a percentage estimated in the literature from 8% to 15% of the normal population.[13] In the case of gonioscopy, we can observe that the extension of the posterior embryotoxon can be greater and be present in the 360○, with a variable thickness of the annulus[12][14][13] and unusually detached and hanging within the anterior chamber.[15] Regarding the iris, we can observe peripheral extensions to Schwalbe's line,[5] which can be thin or thick and extend over the trabecular meshwork, obscuring the scleral spur and even pulling the iris and producing corectopia[6] in iris tissue that can range from atrophy mild stromal to the presence of uveal ectropion, pseudopolycoria or even absence of iris or pseudoacorea.[16] These chamber sinus anomalies predispose half of the cases to open-angle glaucoma, which can manifest throughout life and therefore require regular ophthalmological check-ups.[17][14][18] Other related anomalies are strabismus due to alteration in the insertions of the extraocular muscles or secondary to amblyopia, and with a predisposition to exotropia[17] and retinal detachment.[13][19][20]
Systemic manifestations: In the case of pathologies that affect the extraocular organs, greater attention must be paid to anomalies in the cardiovascular system,[21][22] since they represent the most worrying associations due to their repercussions at the systemic level. These are present in different structures that make it up, such as heart valve defects, the presence of Fallot's tetralogy,[23] atrial septal defects[24][22] or persistent truncus arteriosus.[25] Other alterations described are craniofacial anomalies associated with hypoplasia of the midface, hypertelorism, telecanthus, maxillary hypoplasia, short nasolabial fold, thin upper lip and larger everted lower lip, which are typical facial characteristics although expressed in a variable way.[26] Maxillary hypoplasia and poor tooth development produce a prognathic profile. Inspection of the oral cavity may show microdontia, hypodontia, oligodontia, and a thickened frenulum. The crowns of the anterior teeth may be conical or peg-shaped and the roots may be shortened, the gingival attachments may be reduced, and the enamel may be hypoplastic, contributing to poor dental health.[26][17] There are other described associations such as umbilical,[14][27][28][29] auditory,[30] pituitary,[31][18] psychomotor,[30] size,[32] urethral[29] and anal[33] anomalies as well as albinism.[34]
Pathophysiology
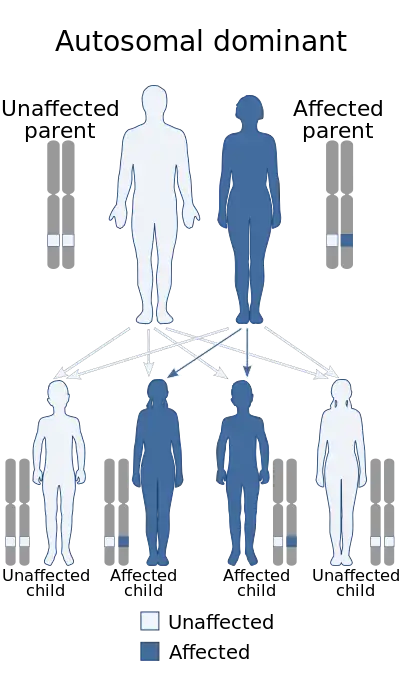
The molecular genetics of Axenfeld–Rieger syndrome are poorly understood, but center on three genes identified by cloning of chromosomal breakpoints from patients.[36][37]
This disorder is inheritable as an autosomal dominant trait,[35] which means the defective gene is located on an autosome, and only one copy of the gene is sufficient to cause the disorder when inherited from a parent who has the disorder. As shown in the diagram, this gives a 50/50 chance of offspring inheriting the condition from an affected parent.
Diagnosis
Although most recognized for its correlation with the onset of glaucoma, the malformation is not limited to the eye, as Axenfeld–Rieger syndrome when associated with the PITX2 genetic mutation usually presents congenital malformations of the face, teeth, and skeletal system.[38]
The most characteristic feature affecting the eye is a distinct corneal posterior arcuate ring, known as an "embryotoxon".[5] In severe cases, iris may be adherent to the cornea anterior to the Schwalbe's line.[39]
One of the three known genetic mutations which cause Rieger syndrome can be identified through genetic samples analysis. About 40% of people with Axenfeld–Rieger have displayed mutations in genes PITX2,[38] FOXC1, and PAX6.[40][41] The difference between Type 1, 2, and 3 Axenfeld–Rieger syndrome is the genetic cause, all three types display the same symptoms and abnormalities.[42]
Classification
The OMIM classification is as follows:
| Type | OMIM | Gene |
|---|---|---|
| Type 1 | 180500 | PITX2 |
| Type 2 | 601499 | possibly FOXO1A[43] |
| Type 3 | 602482 | FOXC1 |
| DeHauwere syndrome | 109120 | Unknown[40] |
Detection of any of these mutations can give patients a clear diagnosis and prenatal procedures such as preimplantation genetic diagnosis, chorionic villus sampling and amniocentesis can be offered to patients and prospective parents.
Management
One of the surgical techniques used to treat this rare disease is the Phakic Retroiridian Pupilloplasty is an original surgical technique involving the creation of a sclerocorneal incision through a peripheral iridotomy, with the surgeon working behind the iris and creating a neopupil with an anterior chamber vitrectome. It requires very few follow-up visits and the patient's recovery is fast.[16]
Eponym
It is named after the German ophthalmologist Theodor Axenfeld[44][5] who studied anterior segment disorders, especially those such as Rieger syndrome and the Axenfeld anomaly.
Axenfeld–Rieger syndrome is characterized by abnormalities of the eyes, teeth, and facial structure.[42] Rieger syndrome, by medical definition, is determined by the presence of malformed teeth, underdeveloped anterior segment of the eyes, and cardiac problems associated with the Axenfeld anomaly.[43] The term "Rieger syndrome" is sometimes used to indicate an association with glaucoma.[38] Glaucoma occurs in up to 50% of patients with Rieger syndrome. Glaucoma develops during adolescence or late childhood, but often occurs in infancy.[5][40] In addition, a prominent Schwalbe's line, an opaque ring around the cornea known as posterior embryotoxon, may arise with hypoplasia of the iris.[36] Below average height and stature, stunted development of the mid-facial features and mental deficiencies may also be observed in patients.[36]
References
- Dhir, L; Frimpong-Ansah, K; Habib, Nabil E (2008). "Missed case of Axenfeld-Rieger syndrome: a case report". Cases Journal. 1 (1): 299. doi:10.1186/1757-1626-1-299. PMC 2585579. PMID 18990239.
- Vieira, Véronique; David, Gabriel; Roche, Olivier; de la Houssaye, Guillaume; Boutboul, Sandrine; Arbogast, Laurence; Kobetz, Alexandra; Orssaud, Christophe; Camand, Olivier; Schorderet, Daniel F.; Munier, Francis; Rossi, Annick; Delezoide, Anne Lise; Marsac, Cécile; Ricquier, Daniel; Dufier, Jean-Louis; Menasche, Maurice; Abitbol, M. (2006). "Identification of four new PITX2 gene mutations in patients with Axenfeld-Rieger syndrome". Molecular Vision. 12: 1448–60. PMID 17167399.
- Fitch, Naomi; Kaback, Martin (1978). "The Axenfeld syndrome and the Rieger syndrome". Journal of Medical Genetics. 15 (1): 30–4. doi:10.1136/jmg.15.1.30. PMC 1012820. PMID 416212.
- Stahl, E. D. (2014). Anterior Segment Dysgenesis. International Ophthalmology Clinics, 54(3), 95–104.
- Axenfeld, T (1920). "Embryotoxon cornea posterius". Klin Monatsbl Augenheilkd. 65: 381–382.
- Rieger H. Contributions to the knowledge of rare malformations of the iris II: hypoplasia of the iris stroma with dislocation and irregularity of the pupil. Albrecht von Graefes arch klin exp ophthalmol. 1935;133:602–635.
- Shields MB. Axenfeld-Rieger syndrome: a theory of mechanism and distinctions from the iridocorneal endothelial syndrome. Trans Am Ophthalmol Soc. 1983;81:736-84.
- Mears AJ, Jordan T, Mirzayans F, et al. Mutaciones del gen forkhead / winged-helix, FKHL7, en pacientes con anomalía de Axenfeld-Rieger. Soy J Hum Genet. 1998; 63 : 1316-1328.
- Nishimura DY, Swiderski RE, Alward WL, et al. El gen del factor de transcripción forkhead FKHL7 es responsable de los fenotipos de glaucoma que se asignan a 6p25. Nat Genet. 1998; 19 : 140-147.
- Semina EV, Reiter R, Leysens NJ, et al. Clonación y caracterización de un nuevo gen del factor de transcripción homeobox relacionado con bicoides, RIEG, implicado en el síndrome de Rieger. Nat Genet. 1996; 14 : 392–399.
- De Hauwere RC, Leroy JG, Adriaenssens K, Van Heule R: Iris dysplasia, orbital hypertelorism, and psychomotor retardation: a dominantly inherited developmental syndrome. J Pediatr 82:679--81, 1973
- Chang, Ta C.; Summers, C. Gail; Schimmenti, Lisa A.; Grajewski, Alana L. (March 2012). "Axenfeld-Rieger syndrome: new perspectives". The British Journal of Ophthalmology. 96 (3): 318–322. doi:10.1136/bjophthalmol-2011-300801. ISSN 1468-2079. PMID 22199394. S2CID 43009007.
- Waring GO 3rd, Rodrigues MM, Laibson PR. Anterior chamber cleavage syndrome. A stepladder classification. Surv Ophthalmol 1975;20:3e27.
- Idrees, F., Vaideanu, D., Fraser, S. G., Sowden, J. C., & Khaw, P. T. (2006). A Review of Anterior Segment Dysgenesis. Survey of Ophthalmology, 51(3), 213–231.
- Espana, E. M., Mora, R., Liebmann, J., & Ritch, R. (2007). Bilateral Prominent Schwalbe Ring in the Anterior Chamber in a Patient with Axenfeld-Rieger Syndrome and Megalocornea. Cornea, 26(3), 379–381.
- Andres, Alza; Eduardo, Galletto (2022). "Pupiloplastia retroiridiana". Oftalmol Clin Exp. 15 (1): e31-e39. ISSN 2718-7446. Retrieved 18 April 2022.
- Chang, T. C., Summers, C. G., Schimmenti, L. A., & Grajewski, A. L. (2011). Axenfeld-Rieger syndrome: new perspectives: Figure 1. British Journal of Ophthalmology, 96(3), 318–322.
- Shields, M. B., Buckley, E., Klintworth, G. K., & Thresher, R. (1985). Axenfeld-Rieger syndrome. A spectrum of developmental disorders. Survey of Ophthalmology, 29(6), 387–409.
- Park SW, Kim HG, Heo H, et al. Anomalous sclera insertion of superior oblique in Axenfeld-Rieger syndrome. Korean J Ophthalmol 2009;23:62e4.
- Spallone A. Retinal detachment in Axenfeld-Rieger syndrome. Br J Ophthalmol 1989;73:559e62
- Antevil J, Umakanthan R, Leacche M, et al. Idiopathic mitral valve disease in a patient presenting with Axenfeld-Rieger syndrome. J Heart Valve Dis 2009;18:349e51.
- Mammi I, De Giorgio P, Clementi M, et al. Cardiovascular anomaly in Rieger syndrome: heterogeneity or contiguity? Acta Ophthalmol Scand 1998;76: 509e12.
- Brear DR & Insler MS (1985): Axenfeld's Syndrome associated with systemic abnormalities. Ann Ophthalmol 17: 291–294.
- Cunningham ET Jr, Eliott D, Miller NR, et al. Familial Axenfeld-Rieger anomaly, atrial septal defect, and sensorineural hearing loss: a possible new genetic syndrome. Arch Ophthalmol 1998;116:78e82.
- Gurbuz-Koz O, Atalay T, Koz C, et al. Axenfeld-Rieger syndrome associated with truncus arteriosus: a case report. Turk J Pediatr 2007;49:444e7
- Jena AK, Kharbanda OP. Axenfeld-Rieger syndrome: report on dental and craniofacial findings. J Clin Pediatr Dent 2005;30:83e8.
- Shields, M. B., Buckley, E., Klintworth, G. K., & Thresher, R. (1985). Axenfeld-Rieger syndrome. A spectrum of developmental disorders. Survey of Ophthalmology, 29(6), 387–409
- Friedman JM: Umbilical dysmorphology. The importance of contemplating the belly button Clin Genet 28:343--7, 1985.
- Jorgenson RJ, Levin LS, Cross HE, et al: The Rieger syndrome. Am J Med Genet 2:307--18, 1978.
- De Hauwere RC, Leroy JG, Adriaenssens K, Van Heule R: Iris dysplasia, orbital hypertelorism, and psychomotor retardation: a dominantly inherited developmental syndrome. J Pediatr 82:679--81, 1973.
- Kleinmann, R. E., Kazarian, E. L., Raptopoulos, V., & Braverman, L. E. (1981). Primary Empty Sella and Rieger's Anomaly of the Anterior Chamber of the Eye. New England Journal of Medicine, 304(2), 90–93.
- Brodsky MC, Whiteside-Michel J, Merin LM: Rieger anomaly and congenital glaucoma in the SHORT syndrome. Arch Ophthalmol 114:1146--7, 1996.
- Crawford RA: Iris dysgenesis with other anomalies. Br J Ophthalmol 51:438--40, 1967.
- Lubin, J. R. (1981). Oculocutaneous Albinism Associated with Corneal Mesodermal Dysgenesis. American Journal of Ophthalmology, 91(3), 347–350.
- "Axenfeld-Rieger syndrome type 1". National Center for Biotechnology Information.
- Suzuki, Katsuhiro; Nakamura, Makoto; Amano, Emi; Mokuno, Kumiko; Shirai, Shoichiro; Terasaki, Hiroko (2006). "Case of chromosome 6p25 terminal deletion associated with Axenfeld–Rieger syndrome and persistent hyperplastic primary vitreous". American Journal of Medical Genetics Part A. 140 (5): 503–8. doi:10.1002/ajmg.a.31085. PMID 16470791. S2CID 30723949.
- Tonoki, Hidefumi; Harada, Naoki; Shimokawa, Osamu; Yosozumi, Ayako; Monzaki, Kadomi; Satoh, Kohei; Kosaki, Rika; Sato, Atsushi; Matsumoto, Naomichi; Iizuka, Susumu (2011). "Axenfeld-Rieger anomaly and Axenfeld-Rieger syndrome: Clinical, molecular-cytogenetic, and DNA array analyses of three patients with chromosomal defects at 6p25". American Journal of Medical Genetics Part A. 155A (12): 2925–32. doi:10.1002/ajmg.a.33858. PMID 22009788. S2CID 520308.
- Meyer-Marcotty, P.; Weisschuh, N.; Dressler, P.; Hartmann, J.; Stellzig-Eisenhauer, A. (2008). "Morphology of the sella turcica in Axenfeld-Rieger syndrome with PITX2 mutation". Journal of Oral Pathology & Medicine. 37 (8): 504–10. doi:10.1111/j.1600-0714.2008.00650.x. PMID 18331556.
- John F., Salmon (2020). "Glaucoma". Kanski's clinical ophthalmology : a systematic approach (9th ed.). Edinburgh: Elsevier. ISBN 978-0-7020-7713-5. OCLC 1131846767.
- Lowry, R. Brian; Gould, Douglas B.; Walter, Michael A.; Savage, Paul R. (2007). "Absence of PITX2, BARX1, and FOXC1 mutations in De Hauwere syndrome (Axenfeld–Rieger anomaly, hydrocephaly, hearing loss): A 25-year follow up". American Journal of Medical Genetics Part A. 143A (11): 1227–30. doi:10.1002/ajmg.a.31732. PMID 17486624. S2CID 44935786.
- Reis, LM; Semina, EV (September 2011). "Genetics of anterior segment dysgenesis disorders". Current Opinion in Ophthalmology. 22 (5): 314–24. doi:10.1097/ICU.0b013e328349412b. PMC 3558283. PMID 21730847.
- "Axenfeld-Rieger syndrome". United States Department of Health and Human Services. November 8, 2016.
- Phillips, Jeffrey C.; del Bono, Elizabeth A.; Haines, Jonathan L.; Pralea, Anca Madalina; Cohen, John S.; Greff, Linda J.; Wiggs, Janey L. (1996). "A second locus for Rieger syndrome maps to chromosome 13q14". American Journal of Human Genetics. 59 (3): 613–9. PMC 1914897. PMID 8751862.
- synd/1284 at Who Named It?
Further reading
- Amendt, Brad A., ed. (2005). The Molecular Mechanisms of Axenfeld-Rieger Syndrome. Medical Intelligence Unit. Springer. doi:10.1007/0-387-28672-1. ISBN 978-0-387-28672-3.
- Agarwal, Sunita; Agarwal, Athiya; Apple, David J., eds. (2002). "Axenfeld-Rieger Syndrome". Textbook of Ophthalmology. Jaypee Brothers. pp. 1049–51. ISBN 978-81-7179-884-1.
- Shields, M.Bruce; Buckley, Edward; Klintworth, Gordon K.; Thresher, Randy (1985). "Axenfeld-Rieger syndrome. A spectrum of developmental disorders". Survey of Ophthalmology. 29 (6): 387–409. doi:10.1016/0039-6257(85)90205-X. PMID 3892740.
- Alward, Wallace L.M. (2000). "Axenfeld-Rieger syndrome in the age of molecular genetics". American Journal of Ophthalmology. 130 (1): 107–15. doi:10.1016/S0002-9394(00)00525-0. PMID 11004268.
External links
- Axenfeld Rieger syndrome at NIH's Office of Rare Diseases
- Axenfeld Rieger anomaly with cardiac defects and sensorineural hearing loss at NIH's Office of Rare Diseases