Börjeson–Forssman–Lehmann syndrome
Börjeson-Forssman-Lehmann syndrome (BFLS) is a rare genetic disease that causes intellectual disability, obesity, and growth defects.[1]
| Börjeson–Forssman–Lehmann syndrome | |
|---|---|
| Other names | Intellectual disability-epilepsy-endocrine disorders syndrome |
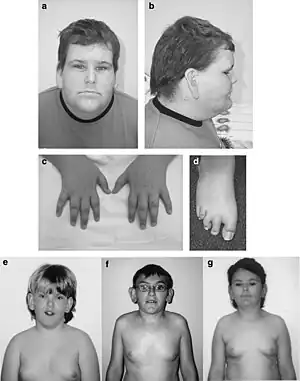 | |
| Affected males with BFLS | |
| Specialty | Medical genetics |
Signs and symptoms
Some symptoms of BFLS are discernible at birth, but they develop over time. Babies with BFLS are born at normal weight but have muscle hypotonia and difficulty feeding. As development progresses, moderate to severe intellectual disability and developmental delays become evident.[2]
Beyond intellectual disability, the central nervous system of affected people shows other symptoms, including impaired vision (cataracts and hyperopia, particularly) and nystagmus.[2][1] Vision impairments can develop before age 30.[1] The peripheral nervous system may also be affected by polyneuropathy.[2] Some individuals may have psychiatric problems, most commonly anxiety disorders, depression, behavioral disorders, and hypersexuality.[3]
The appearance of affected individuals is characteristic, featuring ptosis, large ears, supraorbital ridge, short stature (in approximately half of affected individuals), gynecomastia, deposits of abdominal fat, swollen cheeks and eyelids, short toes, and tapered fingers. Kyphosis or scoliosis may also be present.[1][2][3]
The genitourinary system is also affected by BFLS; the testes of affected children often show hypogonadism and cryptorchidism.[2] Diabetes has co-occurred in several cases.[1]
Hearing loss, epilepsy, cleft lip and palate, acute precursor T-cell acute lymphoblastic leukemia, Legg-Calvé-Perthes disease, and hypopituitarism are uncommon.[2][3]
People with XX chromosomes are usually carriers of the disease and show few, if any symptoms. If affected, they may have obesity, polyneuropathy, or mild intellectual disability.[2]
Pathophysiology
BFLS is an X-linked recessive genetic disease caused by mutations in PHF6, a gene that encodes a zinc finger protein involved in cell growth.[2][1][3] Point mutations lead to less severe forms of the disease than loss of function mutations or deletions.[3] It is highly expressed during development in the pituitary gland, face, and brain.[2] It occurs primarily in people with XY chromosomes because there is only one copy of the X chromosome present and therefore the only copy of PHF6 is the mutated copy.[2]
Diagnosis
Definitive diagnosis of BFLS is made with a genetic test, though it can be suspected where there are several people in a family showing the characteristic symptoms. Magnetic resonance imaging, electroencephalography, and electroneuronography can be used to assess the severity of the disease.[2] Mutations in the PHF6 gene have been shown to be the cause of this condition.[4]
Other diseases that may need to be distinguished from BFLS include Prader–Willi syndrome, Coffin–Lowry syndrome, Klinefelter syndrome, Wilson–Turner syndrome, Bardet–Biedl syndrome, Smith–Fineman–Myers syndrome (Chudley-Lowry syndrome), and Coffin–Siris syndrome.[1][3]
Treatment
There is no cure for BFLS, but its symptoms can be managed with surgery and medication. Surgery is used to treat cryptorchidism and cleft palate, whereas medication is used to treat epilepsy that may result from the condition.[2]
Prognosis
Though affected children can have severe developmental delays, they typically learn to walk between 4 and 6 years old.[2] Intellectual disability in affected individuals does not worsen over time;[3] some affected individuals see their symptoms become less severe.[1]
Epidemiology
BFLS is extremely rare, with less than 100 cases reported in the literature since its discovery. As with other X-linked diseases, it mostly occurs in males, however females can be affected, particularly if they have skewed X-inactivation.[2][5] As of 2022, a patient advocate group BFLS Inc is forming a patient registry.
History
BFLS was described in 1962 by the physicians for whom it is named.[2]
References
- "Börjeson-Forssman-Lehman Syndrome - NORD (National Organization for Rare Disorders)". Retrieved 2015-07-25.
- "Börjeson-Forssman-Lehmann syndrome". www.socialstyrelsen.se. Archived from the original on 2019-04-08. Retrieved 2015-07-24.
- "Orphanet: Borjeson Forssman Lehmann syndrome". Orphanet. Retrieved 2015-07-24.
- Ernst A, Le VQ, Højland AT, Pedersen IS, Sørensen TH, Bjerregaard LL, Lyngbye TJ, Gammelager NM, Krarup H, Petersen MB (2015) The PHF6 mutation c.1A>G; pM1V causes Börjeson-Forsman-Lehmann syndrome in a family with four affected Young boys. Mol Syndromol 6(4):181-186
- Zweier, Christiane; Kraus, Cornelia; Brueton, Louise; Cole, Trevor; Degenhardt, Franziska; Engels, Hartmut; Gillessen-Kaesbach, Gabriele; Graul-Neumann, Luitgard; Horn, Denise; Hoyer, Juliane; Just, Walter; Rauch, Anita; Reis, André; Wollnik, Bernd; Zeschnigk, Michael (2013-12-01). "A new face of Borjeson–Forssman–Lehmann syndrome? De novo mutations in PHF6 in seven females with a distinct phenotype". Journal of Medical Genetics. 50 (12): 838–847. doi:10.1136/jmedgenet-2013-101918. ISSN 0022-2593. PMID 24092917. S2CID 44490262.