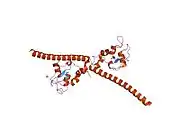
Survivin
Survivin, also called baculoviral inhibitor of apoptosis repeat-containing 5 or BIRC5, is a protein that, in humans, is encoded by the BIRC5 gene.[5][6]





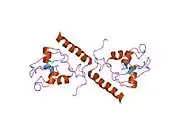
Survivin is a member of the inhibitor of apoptosis (IAP) family. The survivin protein functions to inhibit caspase activation, thereby leading to negative regulation of apoptosis or programmed cell death. This has been shown by disruption of survivin induction pathways leading to increase in apoptosis and decrease in tumour growth. The survivin protein is expressed highly in most human tumours and fetal tissue, but is completely absent in terminally differentiated cells.[7] These data suggest survivin might provide a new target for cancer therapy that would discriminate between transformed and normal cells. Survivin expression is also highly regulated by the cell cycle and is only expressed in the G2-M phase. It is known that Survivin localizes to the mitotic spindle by interaction with tubulin during mitosis and may play a contributing role in regulating mitosis. The molecular mechanisms of survivin regulation are still not well understood, but regulation of survivin seems to be linked to the p53 protein. It also is a direct target gene of the Wnt pathway and is upregulated by beta-catenin.[8]
IAP family of anti-apoptotic proteins
Survivin is a member of the IAP family of antiapoptotic proteins. It is shown to be conserved in function across evolution as homologues of the protein are found both in vertebrates and invertebrates.[9] The first members of the IAPs identified were from the baculovirus IAPs, Cp-IAP and Op-IAP, which bind to and inhibit caspases as a mechanism that contributes to its efficient infection and replication cycle in the host.[9] Later, five more human IAPs that included XIAP, c-IAPl, C-IAP2, NAIP, and survivin were discovered. Survivin, like the others, was discovered by its structural homology to IAP family of proteins in human B-cell lymphoma. The human IAPs, XIAP, c-IAPl, C-IAP2 have been shown to bind to caspase-3 and -7, which are the effector caspases in the signaling pathway of apoptosis.[9] It is not known with absolute certainty though, how the IAPs inhibit apoptosis mechanistically at the molecular level.
A common feature that is present in all IAPs in the presence of a BIR (Baculovirus IAP Repeat, a ~70 amino acid motif) in one to three copies. It was shown by Tamm et al. that knocking out BIR2 from XIAP was enough to cause a loss of function in terms of XIAPs ability to inhibit caspases. This gives the implication that it is within these BIR motifs that contains the anti-apoptotic function of these IAPs. Survivin's one BIR domain shows a similar sequence compared to that of XIAP's BIR domains.[9]
Isoforms
The single survivin gene can give rise to four different alternatively spliced transcripts:[10]
- Survivin, which has a three-intron–four-exon structure in both the mouse and human.
- Survivin-2B, which has an insertion of an alternative exon 2.
- Survivin-Delta-Ex-3, which has exon 3 removed. The removal of exon 3 results in a frame shift that generates a unique carboxyl terminus with a new function. This new function may involve a nuclear localization signal. Moreover, a mitochondrial localization signal is also generated.
- Survivin-3B, which has an insertion of an alternative exon 3.
Structure
A structural feature common to all IAP family proteins is that they all contain at least one baculoviral IAP repeat (BIR) domain characterized by a conserved zinc-coordinating Cys/His motif at the N-terminal half of the protein.[11][12]
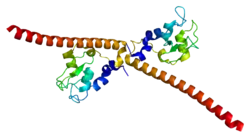
Survivin is distinguished from other IAP family members in that it has only one BIR domain.[11][12] The mice and human BIR domain of survivin are very similar structurally except for two differences that may affect function variability. The human survivin also contains an elongated C-terminal helix comprising 42 amino acids.[11][12] Survivin is 16.5 kDa large and is the smallest member of the IAP family.[11][12] X-ray crystallography has shown two molecules of human survivin coming together to form a bowtie-shape dimer through a hydrophobic interface.[11][12] This interface includes N-terminal residues 6-10 just before the BIR domain region and the 10 residue region connecting the BIR domain to the C-terminal helix.[11][12] The structural integrity of the determined crystal structure of survivin is quite reliable, as physiological conditions were used to obtain the images.
Function
Apoptosis
Apoptosis, the process of programmed cell death, involves complex signaling pathways and cascades of molecular events. This process is needed for proper development during embryonic and fetal growth where there is destruction and reconstruction of cellular structures. In adult organisms, apoptosis is needed to maintain differentiated tissue by striking the balance between proliferation and cell death. It is known that intracellular proteases called caspases degrade the cellular contents of the cell by proteolysis upon activation of the death pathway.
Mammalian cells have two main pathways that lead to apoptosis.
1. Extrinsic pathway: Initiated by extrinsic ligands binding to death receptors on the surface of the cell. An example of this is the binding of tumour necrosis factor-alpha (TNF-alpha) to TNF-alpha receptor. An example of a TNF receptor is Fas (CD95), which recruits activator caspases like caspase-8 upon binding TNF at the cell surface. The activation of the initiator caspases then initiates a downstream cascade of events that results in the induction of effector caspases that function in apoptosis.[9][13]
2. Intrinsic pathway: This pathway is initiated by intracellular or environmental stimuli. It is focused on detecting the improper functioning of the mitochondria in the cell and, as a result, activates signaling pathways to commit suicide. The membrane permeability of the mitochondria increases and particular proteins are released into the cytoplasm that facilitates the activation of initiator caspases. The particular protein released from the mitochondria is cytochrome c. Cytochrome c then binds to Apaf-1 in the cytosol and results in the activation of initiator caspase-9. The activation of the initiator caspases then initiates a downstream cascade of events that results in the induction of effector caspases that function in apoptosis.[9][13]
One family of proteins called IAPs plays a role in regulating cell death by inhibiting the process. IAPs like survivin, inhibit apoptosis by physically binding to and inhibiting proper caspase function.[9] The function of IAPs is evolutionarily conserved as Drosophila homologues of IAPs have been shown to be essential for cell survival.[9]
IAPs have been implicated in studies to have a regulatory effect on cell division. Yeast cells with knock-outs of certain IAP genes did not show problems associated with cell death, but showed defects in mitosis characterized by improper chromosome segregation or failed cytokinesis.[9]
Deletion of particular IAPs does not seem to have a profound effect on the cell-death pathway as there is a redundancy of function by the many IAPs that exist in a cell.[9] They have been implicated, however, to play a role in maintaining an anti-apoptotic environment intracellularly. Changing the expression of particular IAPs has shown an increase in spontaneous cell death induction or increased sensitivity to death stimuli.[9]
Inhibition of Bax and Fas-induced apoptosis
Tamm et al. have shown that survivin inhibits both Bax and Fas-induced apoptotic pathways.[9] The experiment involved transfecting HEK 293 cells with a Bax-encoding plasmid, which resulted in an increase in apoptosis (~7 fold) as measured by DAPI staining.[9] They then contransfected the 293 cells with Bax-encoding plasmid and survivin-encoding plasmids. They observed that cells transfected along with the survivin showed a significant decrease in apoptosis (~3 fold). A similar result also showed for cells transfected with the Fas-overexpressing plasmid. Immunoblots were performed and confirmed that survivin does not inhibit by mechanism of preventing Bax or Fas protein from being made into fully functional proteins.[9] Therefore, survivin should be acting somewhere downstream of the Bax or Fas signaling pathway to inhibit apoptosis through these pathways.[9]
Interaction with caspase-3 and -7
In this part of the experiment, Tamm et al. transfected 293 cells with survivin and lysed them to obtain cell lysate. The lysates were incubated with different caspase forms and survivin was immunopercipitated with anti-survivin antibody. The idea behind this is that, if survivin binds physically with the caspase it is incubated with, it will be co-precipitated along with the survivin while everything else in the lysate is washed away. The immunoprecipitates were then run on SDS-PAGE and then immunoblotted for detection of the desired caspase. If the caspase of interest was detected, it meant that it was bound to survivin in the immunoprecipitation step implicating that survivin and the particular caspase had bound beforehand. Active caspase-3 and -7 coimmunoprecipitated with survivin. The inactive proforms of caspase-3 and -7 did not bind survivin.[9] Survivin also does not bind to active caspase-8.[9] Caspase-3 and -7 are effector proteases whereas caspase-8 is an initiator caspase that sits more upstream in the apoptotic pathway.[9] These results demonstrate survivin's capability to bind with particular caspases in vitro, but may not necessarily translate over to actual physiological conditions. Later, a 2001 study confirmed that human survivin tightly binds caspase-3 and -7 when expressed in E. coli.[14]
Further evidence to support the idea that survivin blocks apoptosis by directly inhibiting caspases was given by Tamm et al. 293 cells were transfected with either overexposed caspase-3 or -7 encoding plasmid and with survivin. They showed that survivin inhibited processing of these two caspases into their active forms. While survivin has been shown as mentioned above to bind to only the active forms of these caspases, it is likely here that survivin inhibits the active forms of the caspases resulting from cleaving and activating more of its own proforms. Thus, survivin acts possibly by preventing such a cascade of cleavage and activation amplification from happening resulting in decreased apoptosis.[9]
In similar manner, looking at the mitochondrial pathway of apoptosis, cytochrome c was transiently expressed in 293 cells to look at the inhibitory effects survivin had on this pathway. Although the details are not here, survivin was shown to also inhibit cytochrome c and caspase-8-induced activation of caspases.[9]
Regulation of cytokinesis
While the mechanism by which survivin may regulate cell mitosis and cytokinesis is not known, the observations made on its localization during mitosis suggests strongly that it is involved in some way in the cytokinetic process.
Proliferating Daoy cells were placed on a glass coverslip, fixed and stained with fluorescent antibodies for survivin and alpha-tubulin. Immunoflourescence using confocal microscopy was used to look at the localization of survivin and tubulin during the cell-cycle to look for any patterns of survivin expression. Survivin was absent in interphase, but present in the G2-M phase.[10]
During the different stages of mitosis, one could see that survivin follows a certain localization pattern. At prophase and metaphase, survivin is mainly nuclear in location.[10] During prophase, as the chromatin condenses so that it is visible under the microscope, survivin starts to move to the centromeres.[10] At prometaphase when the nuclear membrane dissociates and spindle microtubules cross over the nuclear region, survivin stays put at the centromeres.[10] At metaphase, when the chromosomes align at the middle plate and are pulled with high tension to either pole by the kinetochore attachments, survivin then associates with the kinetochores.[10] At anaphase as separation of the chromatids happens, the kinetochore microtubules shorten as the chromosomes move towards to the spindle poles and survivin also moves along to the midplate.[10] Survivin thus accumulates at the midplate at telophase.[10] Finally, survivin localizes to the midbody at the cleavage furrow.[10]
Interaction and localization to the mitochondria
It has been shown that survivin can heterodimerize individually with the two splice variants Survivin-2B and survivin-deltaEx3.[10] Evidence of the heterodimerization of survivin splice variants with survivin was shown with co-immunoprecipitation experiments after cotransfection with the respective survivin variants with survivin. To determine the localization of exogenously expressed survivin-2B and survivin-deltaEx3, fusion constructs of the proteins were made with GFP and HcRed respectively and Daoy cells were transfected with the plasmid constructs. Survivin was also tagged with a fluorescent protein. The fusion of the survivin variants with the fluorescent molecules allows for simple detection of cellular location by fluorescence microscopy. Survivin-2B by itself, localized to both nuclear and cytoplasmic compartments whereas survivin-deltaEx3 localized only in the nucleus.[10] The localization of the three variants (survivin, Survivin-2B, and survivin-deltaEx3) differ, however, when cotransfected together rather than individually.[10]
To see which subcellular compartments contained the survivin splice variants complexes, fluorescent antibody markers for different organelles in the cell were employed. The assumption is that, under fluorescence microscopy, if the particular survivin complex is located in that particular cell compartment, one would observe an overlap from the fluorescence given off by the tagged survivin complex and the tagged compartment as well. Different color fluorescence is used to distinguish compartment from survivin.
- Endoplasmic reticulum and lyosomes: no colocalization
- Mitochondria and golgi: both survivin/survivin-2B and survivin/survivin-deltaEx3 colocalize
To verify these observations, they fractionated the subcellular compartments and performed western blot analysis to definitively say that survivin complexes did indeed localize at these compartments.
Expression in different carcinomas
Survivin is known to be expressed during fetal development and across most tumour cell types, but is rarely present in normal, non-malignant adult cells.[15] Tamm et al. showed that survivin was expressed in all 60 different human tumour lines used in the National Cancer Institute's cancer drug-screening program, with the highest levels of expression in breast and lung cancer lines and the lowest levels in renal cancers.[9] Knowing the relative expression levels of survivin in different tumour types may prove helpful as survivin-related therapy may be administered depending on the expression level and reliance of the tumour type on survivin for resistance to apoptosis.
As an oncogene
Survivin can be regarded as an oncogene as its aberrant overexpression in most cancer cells contributes to their resistance to apoptotic stimuli and chemotherapeutic therapies, thus contributing to their ongoing survival.
Genomic instability
Most human cancers have been found to have gains and losses of chromosomes that may be due to chromosomal instability (CIN). One of the things that cause CIN is the inactivation of genes that control the proper segregation of the sister chromatids during mitosis. In gaining a better understanding of survivin's function in mitotic regulation, scientists have looked into the area of genomic instability. It is known that survivin associates with microtubules of the mitotic spindle at the start of mitosis.[16]
It has been shown in the literature that knocking out survivin in cancer cells will disrupt microtubule formation and result in polyploidy as well as massive apoptosis.[16] It has also been shown that survivin-depleted cells exit mitosis without achieving proper chromosome alignment and then reforms single tetraploid nuclei.[16] Further evidence also suggests that survivin is needed for sustaining mitotic arrest upon encounter with mitosis problems.[16] The evidence mentioned above implicates that survivin plays an important regulatory role both in the progression of mitosis and sustaining mitotic arrest. This seems strange, as survivin is known to be highly upregulated in most cancer cells (that usually contain chromosome instability characteristics), and its function is that which promotes proper regulation of mitosis.
Regulation by p53
p53 inhibits survivin expression at the transcriptional level
Wild-type p53 has been shown to repress survivin expression at the mRNA level.[17] Using an adenovirus vector for wild-type p53, human ovarian cancer cell line 2774qw1 (which expresses mutant p53) was transfected. mRNA levels of survivin were analyzed by real-time quantitative PCR (RT-PCR) and showed time-dependent down regulation of survivin mRNA levels when the cells were infected with wild-type p53.[17] A 3.6 fold decrease of survivin mRNA level was observed 16 hours after infection initiation and decreased 6.7 fold 24 hours after infection.[17] Western blot results do show that there is indeed the p53 from the adenoviral vector was being expressed in the cells using antibody specific for p53. The expression of p53 levels indicative of its role in survivin repression shows that p53 started to be expressed 6 hours into infection and had its highest level at 16–24 hours.[17] To further confirm that endogenous wild-type p53 is really causing the repression of survivin gene expression, the authors induced A549 (human lung cancer cell line with wild-type p53) and T47D (human breast cancer cell line with mutant p53) cells with DNA-damaging agent adriamycin to trigger the physiological p53 apoptotic response in these cancer cells and compare the survivin levels measured to the same cells without DNA damage induction. The A549 line, which intrinsically has functioning wild-type p53, showed significant reduction in survivin levels compared to non-induced cells.[17] This same effect was not seen in T47D cells that carry mutant inactive p53.[17]
P53's normal function is to regulate genes that control apoptosis. As survivin is a known inhibitor of apoptosis, it can be implied that p53 repression of survivin is one mechanism by which cells can undergo apoptosis upon induction by apoptotic stimuli or signals. When survivin is over-expressed in the cell lines mentioned in the previous paragraph, apoptotic response from DNA-damaging agent adriamycin decreased in a dose-dependent manner.[17] This suggests that down-regulation of survivin by p53 is important for p53-mediated apoptotic pathway to successfully result in apoptosis. It is known that a defining characteristic of most tumors is the over-expression of survivin and the complete loss of wild-type p53.[17] The evidence put forth by Mirza et al. shows that there exists a link between survivin and p53 that can possibly explain a critical event that contributes to cancer progression.
p53 suppression of survivin expression
In order to see whether p53 re-expression in cancer cells (that have lost p53 expression) has the suppressive effect on the promoter of the survivin gene, a luciferase reporter construct was made. The isolated survivin promoter was placed upstream of the luciferase reporter gene. In a luciferase reporter assay, if the promoter is active, the luciferase gene is transcribed and translated into a product that gives off light that can measured quantitatively and, thus, represents the activity of the promoter. This construct was transfected into cancer cells that had either wild-type or mutant p53. High luciferase activity was measured in the cells with mutant p53 and significantly lower luciferase levels were measured for cells with wild-type p53.[17]
Transfection of different cell types with wild-type p53 was associated with a strong repression of the survivin promoter.[17] Transfection with mutant p53 was not shown to strongly repress the survivin promoter.[17] More luciferase constructs were prepared with varying degrees of deletion from the 5' end of the survivin promoter region. At one point, there was deletion that caused the survivin levels to be indifferent to the presence of the p53 over-expression plasmid, indicating that there is a specific region proximal to the transcription start site that is needed for p53 suppression of survivin.[17] Although it has been found that two p53 binding sites are located on the survivin gene promoter, analysis using deletions and mutations has shown that these sites are not essential to transcriptional inactivation.[17]
Instead, it is observed that modification of the chromatin inside of the promoter region may be responsible for the transcriptional repression of the survivin gene. This is explained below in the epigenetic regulation section.[17]
Cell cycle regulation
Survivin is shown to be clearly regulated by the cell cycle, as its expression is found to be dominant only in the G2/M phase.[13] This regulation exists at the transcriptional level, as there is evidence of the presence of cell-cycle-dependent element/cell-cycle gene homology region (CDE/CHR)boxes located in the survivin promoter region.[13] Further evidence to support this mechanism of regulation includes the evidence that surivin is poly-ubiquinated and degraded by proteasomes during interphase of the cell cycle.[13] Moreover, survivin has been shown to localize to components of the mitotic spindle during metaphase and anaphase of mitosis.[13] Physical association between polymerized tubulin and survivin have been shown in vitro as well.[13] It is also shown that post-transcriptional modification of survivin involving the phosphorylation of Thr34 leads to increased protein stability in the G2/M phase of the cell cycle.[13]
It is known from Mirza et al. that repression of survivin by p53 is not a result of any cell cycle progressive regulation. The same experiment by Mirza et al. with regard to determining p53 suppression of survivin at the transcriptional level was repeated, but this time for cells arrested in different stages of the cell cycle. It was shown that, although p53 arrests the numbers of cells to different extents in different phases, the measured level of survivin mRNA and protein levels were the same across all the samples transfected with the wild-type p53. This shows that p53 acts in a cell-cycle independent manner to inhibit survivin expression.[17]
Epigenetic and genetic regulation
As observed through the literature, survivin is found to be over-expressed across many tumour types. Scientists are not sure of the mechanism that causes this abnormal over-expression of survivin; however, p53 is downregulated in almost all cancers, so it is tempting to suggest that survivin over-expression is due to p53 inactivity. Wagner et al. investigated the possible molecular mechanism involved with the over expression of survivin in acute myeloid leukemia (AML). In their experiments, they did both an epigenetic and a genetic analysis of the survivin gene promoter region in AML patients and compared the observations to what was seen in peripheral blood mononuclear cells (PBMCs) that have been shown to express no survivin. Assuming that the molecular mechanism of survivin re-expression in cancerous cells is at the transcriptional level, the authors decided to look at particular parts of the promoter region of survivin in order to see what happens in cancer cells that does not happen in normal cells that causes such a high level of survivin to be expressed. With regards to an epigenetic mechanism of survivin gene regulation, the authors measured the methylation status of the survivin promoter, since it is accepted that methylation of genes plays an important role in carcinogenesis by silencing of certain genes or vice versa. The authors used methylation specific polymerase chain reaction with bisulfite sequencing methods to measure the promoter methylation status in AML and PBMCs and found unmethylated survivin promoters in both groups.[18] This result shows that DNA methylation status is not an important regulator of survivin re-expression during leukemogenesis.[18] However, De Carvalho et al. performed a DNA methylation screening and identified that DNA methylation of IRAK3 plays a key role in survivin up-regulation in different types of Cancer,[19] suggesting that epigenetic mechanisms plays an indirect role on abnormal over-expression of survivin. With regard to genetic analysis of the survivin promoter region, the isolated DNA of AML and PBMCs were treated with bisulfite, and the survivin promoter region sequence was amplified out with PCR and sequenced to look for any particular genetic changes in the DNA sequence between the two groups. Three single-nucleotide polymorphisms (SNPs) were identified and were all present both in AML patients and in healthy donors. This result suggests that the occurrence of these SNPs in the promoter region of the survivin gene also appears to be of no importance to survivin expression.[18] However, it has not been ruled out yet that there may be other possible epigenetic mechanisms that may be responsible for a high level of survivin expression observed in cancer cells and not in normal cells. For example, the acetylation profile of the survivin promoter region can also be looked at. Different cancer and tissue types may have slight or significant differences in the way survivin expression is regulated in the cell, and, thus, the methylation status or genetic differences in the survivin promoter may be observed to be different in different tissues. Thus, further experiments assessing the epigenetic and genetic profile of different tumour types must be investigated.
As a drug target
Expression in cancer as a tool for cancer-directed therapy
Survivin is known to be highly expressed in most tumour cell types and absent in normal cells, making it a good target for cancer therapy.[20][21][22][23][24] The exploitation of survivin's over-active promoter in most cancer cell types allows for the delivery of therapeutics only in cancer cells and removed from normal cells.[25]
Small interfering RNA (siRNA) are synthetic antisense oligonucleotides to the mRNA of the gene of interest that works to silence the expression of a particular gene by its complementary binding. siRNAs, such as LY2181308, bound to the respective mRNA results in disruption of translation of that particular gene and thus the absence of that protein in the cell. Thus, the use of siRNAs has great potential to be a human therapeutic, as it can target and silence the expression of potentially any protein you want. A problem arises when siRNA expression in a cell cannot be controlled, allowing its constitutive expression to cause toxic side-effects. With regard to practical treatment of cancer, it is required to either deliver the siRNAs specifically into cancer cells or control the siRNA expression. Previous methods of siRNA therapy employ the use of siRNA sequences cloned into vectors under the control of constitutively active promoters.[25] This causes a problem, as this model is non-specific to cancer cells and damages normal cells too.[25] Knowing that survivin is over-expressed specifically in cancer cells and absent in normal cells, one can imply that the survivin promoter is active only in cancer cells. Thus, the exploitation of this difference between cancer cells and normal cells will allow appropriate therapy directed only at the cells in a patient that are harmful. In an experiment to demonstrate this idea, Trang et al. have created a cancer-specific vector expressing siRNA for green fluorescent protein (GFP) under the human survivin promoter. MCF7 breast cancer cells were cotransfected with this vector and a GFP-expressing vector as well. Their major finding was that MCF7 cells transfected with the siRNA vector for GFP under the survivin promoter had a significant reduction in GFP expression then the cells transfected with the siRNA vector under a cancer non-specific promoter.[25] Moreover, normal non-cancerous cells transfected in the same way mentioned above showed no significant reduction in GFP expression.[25] This is implying that, in normal cells, survivin promoter is not active, and, thus, the siRNA will not be expressed under an inactive survivin promoter.[25]
Antisense oligonucleotides targeting survivin mRNA
As it is known that survivin is over-expressed in most cancers, which may be contributing to the cancer cells' resistance to apoptotic stimuli from the environment. The use of antisense survivin therapy hopes to render cancer cells susceptible to apoptosis by eliminating survivin expression in the cancer cells.[8]
Olie et al. developed different 20-mer phosphorothioate antisense oligonucleotides that target different regions in the mRNA of the survivin gene. The antisense function of the oligonucleotides allows binding to surviving mRNA and, depending on the region on which it binds, might inhibit surviving mRNA from being translated into a functional protein. Real-time PCR was used to assess the levels of mRNA present in a lung adenocarcinoma cell line A549 that overexpresses survivin. The best antisense oligonucleotide was identified that effectively down-regulated survivin mRNA levels and resulted in apoptosis of the cells. Survivin's role in cancer development in the context of a signaling pathway is its ability to inhibit activation of downstream caspase-3 and -7 from apoptosis inducing stimuli. The overexpression of survivin in tumors may serve to increase the tumors resistance to apoptosis and, thus, contribute to cell immortality even in the presence of death stimuli.[25] In this experiment, the oligonucleotide 4003 that targets nucleotides 232-251 of survivin mRNA was found to be the most effective at down-regulating the levels of survivin mRNA in the A549 tumour line.[25] The 4003 oligonucleotides were introduced into the tumour cells by transfection. Further experiments were then conducted on 4003. One of the additional experiments involved determining the dose-dependent effect of 4003 on the down-regulation of survivin mRNA levels. It was found that a concentration of 400 nM resulted in a maximum down-regulation of 70% of the initial survivin mRNA present.[25] Another experiment on 4003 involved assessing any biological or cytotoxic effect 4003 down-regulation of survivin mRNA has on A549 cells using the MTT assay. The numbers of A549 cells transfected with 4003 significantly decreased with increasing concentration of 4003 compared to cells transfected either with a mismatch form of the 4003 or lipofectin control.[25] Many physical observations that confirmed the induction of apoptosis by 4003 were made. For example, lysates of the 4003-treated cells showed increased levels of caspase-3-like protease activity; nuclei were observed to be condensed and chromatin was fragmented.
Cancer immunotherapy
Survivin has been a target of attention in recent years for cancer immunotherapy, as it is an antigen that is expressed mostly in cancer cells and absent in normal cells. This is because survivin is deemed to be a crucial player in tumour survival. There has been much evidence accumulated over the years that shows survivin as a strong T-cell-activating antigen, and clinical trials have already been initiated to prove its usefulness in the clinic.[26]
Activation of the adaptive immune system
A. Cellular T cell response
The first evidence of survivin-specific CTL recognition and killing was shown in an assay wherein cytotoxic T cells (CTLs) induced lysis of B cells transfected to present survivin peptides on its surface.[26] The naive CD8+ T cells were primed with dendritic cells and could therefore recognize the specific peptides of survivin presented on the surface Major Histocompatibility Complex I (MHC I) molecules of the B cells.
B. Humoral antibody response
Taking blood samples from cancer patients, scientists have found antibodies that are specific for survivin.[26] These antibodies were absent in the blood samples of healthy normal patients.[26] Therefore, this shows that survivin is able to elicit a full humoral immune response. This may prove useful, as one could measure the level of survivin-specific antibodies in the patient's blood as a monitor of tumour progression.[26] In acquiring the humoral response to tumour antigens such as survivin, CD4+ T cells are activated to induce B cells to produce antibodies directed against the particular antigens.
The isolation of the antibodies specific for survivin peptides is useful, as one can look at the structure and sequence of the epitope binding groove of the antibody and, therefore, deduce possible epitopes that may fit in that particular antibody groove.[26] Therefore, one can determine the particular peptide portion of the survivin protein that is bound most efficiently and most commonly by humoral antibodies generated against survivin. This will lead to the production of more specific survivin vaccines that contain a specific portion of the survivin protein that is known to elicit a good immune response, generate immune memory, and allow for protection from tumour development.
Over-expression in tumours and metastatic tissues
Xiang et al. found a new approach in inhibiting tumour growth and metastasis by simultaneously attacking both the tumour and its vasculature by a cytotoxic T cell (CTL) response against the survivin protein, which will later result in the activation of apoptosis in tumour cells.[27]
The idea and general principle behind his technique is described below. Mice were immunized with the oral vaccination and then subjected to tumour challenges by injecting them in the chest with a certain number of tumour cells and a Matrigel pre-formed extracellular matrix to hold the tumour cells together. The mice were sacrificed and the endothelium tissue was stained with a fluorescent dye that would aid in the quantification of tumour neovascularisation using a Matrigel assay. There was found to be a significant difference between the control and test groups, whereby mice given the vaccine had less angiogenesis from the tumour challenge than the control mice that were not given any of the vaccine prior to tumour challenge.[27] In vitro assays and other tests were also performed to validate the idea of the occurrence of an actual immune response to support what they observed in the mice.[27] For example, the spleen on the challenged mice were isolated and measured for the presence of any cytokines, and specifically activated immune cell groups that would indicative that a specific immune response did occur upon vaccination. The isolated CTLs specific for the survivin protein after vaccination of the mice were used in cytoxicity assays where mice tumour cells expressing survivin were shown to be killed upon incubation with the specific CTLs.[27]
By using an oral DNA vaccine carried in an attenuated non-virulent form of Salmonella typhimurium, which co-encoded secretory chemokine CCL21 and survivin protein in C57BL/6J mice, Xiang et al. have been able to elicit an immune response carried out by dendritic cells (DCs) and CTLs to eliminate and suppress the pulmonary metastases of non-small cell lung carcinoma. The activation of the immune response is most likely taking place in the secondary lymphoid organ called the Peyer's Patch in the small intestine where DCs take up the survivin protein by phagocytosis and present them on their surface receptors to naive CD8+ T cells (uninactivated CTL) to achieve a specific immune response targeting survivin exclusively.[27] Activated CTLs specific for a particular antigen kill their target cells by first recognizing parts of the survivin protein expressed on MHC I (immunohistocompatability) proteins presented on the surface of tumour cells and vasculature and then releasing granules that induce the tumour cells to undergo apoptosis. The DNA vaccine contained the CCL21 secretory chemokine as a way to enhance the likelihood of eliciting the immune response by better mediating the physical interaction of the antigen-presenting DCs and the naive CD8+ T cells, resulting in a greater likelihood of immune activation.[27]
Resveratrol-mediated sensitization
It has been shown by Fulda et al. that the naturally occurring compound resveratrol (a polyphenol found in grapes and red wine) can be used as a sensitizer for anticancer drug-induced apoptosis by the action of causing cell cycle arrest.[28] This cell cycle arrest causes a dramatic decline in survivin levels in the cells, as it is known from the literature that survivin expression is highly linked with the cell cycle phase state. Thus, the decrease in survivin, which is a contributing factor to chemotherapy resistance and apoptosis induction therapies, would render the cancer cells more prone to such cancer treatments. Fulda et al. have demonstrated the benefits of resveratrol through a series of experiments. First, the authors of the paper tested the intrinsic cytotoxic effects of resveratrol. They found that it induced moderate apoptosis levels only in SHEP neuroblastoma cells.[28] After, they tested resveratrol in combination with several different known anticancer agents. They found a consistent increase in the level of apoptosis induced by the drugs when resveratrol was also present.[28] Moreover, they varied the order with which either the drugs or resveratrol was introduced to the cancer cells to determine whether the sequence of treatment had any important effect. It was found that the highest levels of apoptosis induction were observed when resveratrol was added prior to anticancer drug treatment.[28] Next, the authors tested for any differential sensitivity to apoptosis linked to the phase of the cell cycle the cells were in. Analysis by flow cytometry revealed an accumulation of cells in S phase upon treatment with resveratrol. The cells were also halted in different phases of the cell cycle using special compounds and then treated with the anticancer drugs. They found that cells halted in S phase were significantly more sensitive to the cytotoxic effects of the drugs.[28]
To determine the involvement of survivin in resveratrol-mediated sensitization, the authors decided to test whether downregulation of the specific survivin protein expression would confer a similar effect on the phenotype of resveratrol-treated cells. In terms of seeing at which level resveratrol worked, they did a northern blot and found that resveratrol treatment resulted in a decrease in survivin mRNA levels,[28] thus implying resveratrol's inhibitory action at the transcriptional level. To further see whether survivin played a key role in sensitization of the cancer cells to cytotoxic drugs, survivin antisense oligonucleotides were used to knock down any survivin mRNA, and, thus, its possibility to be translated is also eliminated. siRNAs for survivin are complements in sequence to the mRNA sequence encoding survivin. When these siRNAs for survivin are introduced into cells, they will bind to the respective complementary mRNA and, thus, prevent its translation since the mRNA is now impeded from proper physical interaction with the translational machinery. In this way, the siRNAs for survivin effectively downregulates survivin expression level in the cell. Cells treated with antisense oligonucleotides for survivin showed similar sensitization to cytotoxic drugs as cells treated with resveratrol, which offers support for the mechanism of action of resveratrol.[28]
Prostate cancer
It has been observed that the development of hormone resistance in prostate cancer may be due to the upregulation of antiapoptotic genes, one of which is survivin.[29]
Zhang et al. hypothesize that, if survivin is a significant contributor to the development of hormonal therapy resistance in prostate cancer cells, targeting survivin and blocking it would enhance prostate cancer cell susceptibility to anti-androgen therapy. (Anti-androgen therapy uses drugs to eliminate the presence of androgens in the cell and cellular environment, since such androgens are known to enhance tumour immortality in prostate cancer cells.) Zhang et al. first assessed the level of survivin expression of LNCaP (an androgen-dependent prostate cancer cell line that expresses intact androgen receptors) using quantitative Western analysis and found high expression of survivin in these cells.[29] Cells exposed to dihydrotestosterone (DHT) showed increased levels of survivin expression only and not other IAP family members.[29] This result suggests that androgens may upregulate survivin, which contributes to the resistance to apoptosis observed in the tumour cells.[29] Next, with the addition of flutamide (an antiandrogen) to the cells, survivin levels were observed to significantly decrease.[29] The LNCaP cells were transduced separately with the different constructs of the survivin gene (mutant or wild-type) and subjected to flutamide treatment and assessed for the apoptosis level. Flutamide-treated survivin mutant-transduced cells were shown to significantly increase apoptosis by double that of flutamide treatment alone.[29] On the other end, overexpression of the wild-type survivin was found to significantly reduce the apoptosis levels from flutamide treatment compared to flutamide treatment alone.[29] Therefore, these results support the hypothesis that survivin plays a role in the anti-apoptotic nature of the LNCaP cancer cell line and that inhibiting survivin in prostate cancer cells appears to enhance the therapeutic effect of flutamide.
Interactions
Survivin has been shown to interact with:
References
- GRCh38: Ensembl release 89: ENSG00000089685 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000017716 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Altieri DC (February 1994). "Molecular cloning of effector cell protease receptor-1, a novel cell surface receptor for the protease factor Xa". J. Biol. Chem. 269 (5): 3139–42. doi:10.1016/S0021-9258(17)41838-2. PMID 8106347.
- Altieri DC (November 1994). "Splicing of effector cell protease receptor-1 mRNA is modulated by an unusual retained intron". Biochemistry. 33 (46): 13848–55. doi:10.1021/bi00250a039. PMID 7947793.
- Sah NK, Khan Z, Khan GJ, Bisen PS (December 2006). "Structural, functional and therapeutic biology of survivin". Cancer Lett. 244 (2): 164–71. doi:10.1016/j.canlet.2006.03.007. PMID 16621243.
- Olie RA, Simões-Wüst AP, Baumann B, Leech SH, Fabbro D, Stahel RA, Zangemeister-Wittke U (June 2000). "A novel antisense oligonucleotide targeting survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy". Cancer Res. 60 (11): 2805–9. PMID 10850418.
- Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T, Reed JC (December 1998). "IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs". Cancer Res. 58 (23): 5315–20. PMID 9850056.
- Caldas H, Jiang Y, Holloway MP, Fangusaro J, Mahotka C, Conway EM, Altura RA (March 2005). "Survivin splice variants regulate the balance between proliferation and cell death". Oncogene. 24 (12): 1994–2007. doi:10.1038/sj.onc.1208350. PMID 15688031.
- Verdecia MA, Huang H, Dutil E, Kaiser DA, Hunter T, Noel JP (July 2000). "Structure of the human anti-apoptotic protein surviving reveals a dimeric arrangement". Nat. Struct. Biol. 7 (7): 602–8. doi:10.1038/76838. PMID 10876248. S2CID 30730657.
- Chantalat L, Skoufias DA, Kleman JP, Jung B, Dideberg O, Margolis RL (July 2000). "Crystal structure of human survivin reveals a bow tie-shaped dimer with two unusual alpha-helical extensions". Mol. Cell. 6 (1): 183–9. doi:10.1016/s1097-2765(00)00019-8. PMID 10949039.
- Altieri DC (January 2003). "Validating survivin as a cancer therapeutic target". Nat. Rev. Cancer. 3 (1): 46–54. doi:10.1038/nrc968. PMID 12509766. S2CID 8567453.
- Shin S, Sung BJ, Cho YS, Kim HJ, Ha NC, Hwang JI, Chung CW, Jung YK, Oh BH (January 2001). "An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and -7". Biochemistry. 40 (4): 1117–23. doi:10.1021/bi001603q. PMID 11170436.
- Ambrosini G, Adida C, Altieri DC (1997). "A novel anti-apoptotic gene, survivin, expressed in cancer and lymphoma". Nat. Med. 3 (8): 917–21. doi:10.1038/nm0897-917. PMID 9256286. S2CID 3062648.
- Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G (April 2004). "Cell death by mitotic catastrophe: a molecular definition". Oncogene. 23 (16): 2825–37. doi:10.1038/sj.onc.1207528. PMID 15077146.
- Mirza A, McGuirk M, Hockenberry TN, Wu Q, Ashar H, Black S, Wen SF, Wang L, Kirschmeier P, Bishop WR, Nielsen LL, Pickett CB, Liu S (April 2002). "Human survivin is negatively regulated by wild-type p53 and participates in p53-dependent apoptotic pathway". Oncogene. 21 (17): 2613–22. doi:10.1038/sj.onc.1205353. PMID 11965534.
- Wagner M, Schmelz K, Dörken B, Tamm I (July 2008). "Epigenetic and genetic analysis of the survivin promoter in acute myeloid leukemia". Leuk. Res. 32 (7): 1054–60. doi:10.1016/j.leukres.2007.11.013. PMID 18206228.
- De Carvalho DD, Sharma S, You JS, Su SF, Taberlay PC, Kelly TK, Yang X, Liang G, Jones PA (May 2012). "DNA methylation screening identifies driver epigenetic events of cancer cell survival". Cancer Cell. 21 (5): 655–67. doi:10.1016/j.ccr.2012.03.045. PMC 3395886. PMID 22624715.
- Zaffaroni N, Pennati M, Daidone MG (2005). "Survivin as a target for new anticancer interventions". J. Cell. Mol. Med. 9 (2): 360–72. doi:10.1111/j.1582-4934.2005.tb00361.x. PMC 6740253. PMID 15963255.
- Altieri DC (March 2006). "Targeted therapy by disabling crossroad signaling networks: the survivin paradigm". Mol. Cancer Ther. 5 (3): 478–82. doi:10.1158/1535-7163.MCT-05-0436. PMID 16546961.
- Pennati M, Folini M, Zaffaroni N (June 2007). "Targeting survivin in cancer therapy: fulfilled promises and open questions". Carcinogenesis. 28 (6): 1133–9. doi:10.1093/carcin/bgm047. PMID 17341657.
- Mita AC, Mita MM, Nawrocki ST, Giles FJ (August 2008). "Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics". Clin. Cancer Res. 14 (16): 5000–5. doi:10.1158/1078-0432.CCR-08-0746. PMID 18698017.
- Pennati M, Folini M, Zaffaroni N (April 2008). "Targeting survivin in cancer therapy". Expert Opin. Ther. Targets. 12 (4): 463–76. doi:10.1517/14728222.12.4.463. PMID 18348682. S2CID 84568177.
- Huynh T, Wälchli S, Sioud M (December 2006). "Transcriptional targeting of small interfering RNAs into cancer cells". Biochem. Biophys. Res. Commun. 350 (4): 854–9. doi:10.1016/j.bbrc.2006.09.127. PMID 17034763.
- Friedrichs B, Siegel S, Andersen MH, Schmitz N, Zeis M (June 2006). "Survivin-derived peptide epitopes and their role for induction of antitumor immunity in hematological malignancies". Leuk. Lymphoma. 47 (6): 978–85. doi:10.1080/10428190500464062. PMID 16840186. S2CID 27915488.
- Xiang R, Mizutani N, Luo Y, Chiodoni C, Zhou H, Mizutani M, Ba Y, Becker JC, Reisfeld RA (January 2005). "A DNA vaccine targeting survivin combines apoptosis with suppression of angiogenesis in lung tumor eradication". Cancer Res. 65 (2): 553–61. doi:10.1158/0008-5472.553.65.2. PMID 15695399. S2CID 12221226.
- Fulda S, Debatin KM (September 2004). "Sensitization for anticancer drug-induced apoptosis by the chemopreventive agent resveratrol". Oncogene. 23 (40): 6702–11. doi:10.1038/sj.onc.1207630. PMID 15273734.
- Zhang M, Latham DE, Delaney MA, Chakravarti A (April 2005). "Survivin mediates resistance to antiandrogen therapy in prostate cancer". Oncogene. 24 (15): 2474–82. doi:10.1038/sj.onc.1208490. PMID 15735703.
- Wheatley SP, Carvalho A, Vagnarelli P, Earnshaw WC (June 2001). "INCENP is required for proper targeting of Survivin to the centromeres and the anaphase spindle during mitosis". Curr. Biol. 11 (11): 886–90. doi:10.1016/s0960-9822(01)00238-x. PMID 11516652. S2CID 381637.
- Chen J, Jin S, Tahir SK, Zhang H, Liu X, Sarthy AV, McGonigal TP, Liu Z, Rosenberg SH, Ng SC (January 2003). "Survivin enhances Aurora-B kinase activity and localizes Aurora-B in human cells". J. Biol. Chem. 278 (1): 486–90. doi:10.1074/jbc.M211119200. PMID 12419797.
- Sampath SC, Ohi R, Leismann O, Salic A, Pozniakovski A, Funabiki H (July 2004). "The chromosomal passenger complex is required for chromatin-induced microtubule stabilization and spindle assembly". Cell. 118 (2): 187–202. doi:10.1016/j.cell.2004.06.026. PMID 15260989. S2CID 17795816.
- Gassmann R, Carvalho A, Henzing AJ, Ruchaud S, Hudson DF, Honda R, Nigg EA, Gerloff DL, Earnshaw WC (July 2004). "Borealin: a novel chromosomal passenger required for stability of the bipolar mitotic spindle". J. Cell Biol. 166 (2): 179–91. doi:10.1083/jcb.200404001. PMC 2172304. PMID 15249581.
- Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T, Reed JC (December 1998). "IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs". Cancer Res. 58 (23): 5315–20. PMID 9850056.
- Song Z, Yao X, Wu M (June 2003). "Direct interaction between survivin and Smac/DIABLO is essential for the anti-apoptotic activity of survivin during taxol-induced apoptosis". J. Biol. Chem. 278 (25): 23130–40. doi:10.1074/jbc.M300957200. PMID 12660240.
Further reading
- Colnaghi R, Connell CM, Barrett RM, Wheatley SP (November 2006). "Separating the anti-apoptotic and mitotic roles of survivin". J. Biol. Chem. 281 (44): 33450–6. doi:10.1074/jbc.C600164200. PMID 16950794.
- O'Driscoll L, Linehan R, Clynes M (2003). "Survivin: role in normal cells and in pathological conditions" (PDF). Current Cancer Drug Targets. 3 (2): 131–52. doi:10.2174/1568009033482038. hdl:2262/78955. PMID 12678716.
- Chiou SK, Jones MK, Tarnawski AS (2003). "Survivin - an anti-apoptosis protein: its biological roles and implications for cancer and beyond". Med. Sci. Monit. 9 (4): PI25–9. PMID 12709681.
- Ouhtit A, Matrougui K, Bengrine A, Koochekpour S, Zerfaoui M, Yousief Z (2007). "Survivin is not only a death encounter but also a survival protein for invading tumor cells". Front. Biosci. 12: 1260–70. doi:10.2741/2144. PMID 17127378.
- Pennati M, Folini M, Zaffaroni N (2007). "Targeting survivin in cancer therapy: fulfilled promises and open questions". Carcinogenesis. 28 (6): 1133–9. doi:10.1093/carcin/bgm047. PMID 17341657.
- Knauer SK, Mann W, Stauber RH (2007). "Survivin's dual role: an export's view". Cell Cycle. 6 (5): 518–21. doi:10.4161/cc.6.5.3902. PMID 17361097.
- Wang TT, Qian XP, Liu BR (2007). "Survivin: potential role in diagnosis, prognosis and targeted therapy of gastric cancer". World J. Gastroenterol. 13 (20): 2784–90. doi:10.3748/wjg.v13.i20.2784. PMC 4395628. PMID 17569112.
- Stauber RH, Mann W, Knauer SK (2007). "Nuclear and cytoplasmic survivin: molecular mechanism, prognostic, and therapeutic potential". Cancer Res. 67 (13): 5999–6002. doi:10.1158/0008-5472.CAN-07-0494. PMID 17616652.
- Bokarewa M, Tarkowski A, Magnusson M (2007). "Pathological survivin expression links viral infections with pathogenesis of erosive rheumatoid arthritis". Scand. J. Immunol. 66 (2–3): 192–8. doi:10.1111/j.1365-3083.2007.01977.x. PMID 17635796.
- Wolanin K, Piwocka K (2007). "[Role of survivin in mitosis]". Postepy Biochem. 53 (1): 10–8. PMID 17718383.
- Altieri DC (1994). "Splicing of effector cell protease receptor-1 mRNA is modulated by an unusual retained intron". Biochemistry. 33 (46): 13848–55. doi:10.1021/bi00250a039. PMID 7947793.
- Altieri DC (1994). "Molecular cloning of effector cell protease receptor-1, a novel cell surface receptor for the protease factor Xa". J. Biol. Chem. 269 (5): 3139–42. doi:10.1016/S0021-9258(17)41838-2. PMID 8106347.
- Maruyama K, Sugano S (1994). "Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides". Gene. 138 (1–2): 171–4. doi:10.1016/0378-1119(94)90802-8. PMID 8125298.
- Suzuki Y, Yoshitomo-Nakagawa K, Maruyama K, Suyama A, Sugano S (1997). "Construction and characterization of a full length-enriched and a 5'-end-enriched cDNA library". Gene. 200 (1–2): 149–56. doi:10.1016/S0378-1119(97)00411-3. PMID 9373149.
- Ambrosini G, Adida C, Sirugo G, Altieri DC (1998). "Induction of apoptosis and inhibition of cell proliferation by survivin gene targeting". J. Biol. Chem. 273 (18): 11177–82. doi:10.1074/jbc.273.18.11177. PMID 9556606.
- Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T, Reed JC (1998). "IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs". Cancer Res. 58 (23): 5315–20. PMID 9850056.
- Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC (1999). "Control of apoptosis and mitotic spindle checkpoint by survivin". Nature. 396 (6711): 580–4. doi:10.1038/25141. PMID 9859993. S2CID 4329354.
- Mahotka C, Wenzel M, Springer E, Gabbert HE, Gerharz CD (2000). "Survivin-deltaEx3 and survivin-2B: two novel splice variants of the apoptosis inhibitor survivin with different antiapoptotic properties". Cancer Res. 59 (24): 6097–102. PMID 10626797.
- Suzuki A, Ito T, Kawano H, Hayashida M, Hayasaki Y, Tsutomi Y, Akahane K, Nakano T, Miura M, Shiraki K (2000). "Survivin initiates procaspase 3/p21 complex formation as a result of interaction with Cdk4 to resist Fas-mediated cell death". Oncogene. 19 (10): 1346–53. doi:10.1038/sj.onc.1203429. PMID 10713676.
- Verdecia MA, Huang H, Dutil E, Kaiser DA, Hunter T, Noel JP (2000). "Structure of the human anti-apoptotic protein survivin reveals a dimeric arrangement". Nat. Struct. Biol. 7 (7): 602–8. doi:10.1038/76838. PMID 10876248. S2CID 30730657.
PDB gallery | |
|---|---|
|