BLAT (bioinformatics)
BLAT (BLAST-like alignment tool) is a pairwise sequence alignment algorithm that was developed by Jim Kent at the University of California Santa Cruz (UCSC) in the early 2000s to assist in the assembly and annotation of the human genome.[1] It was designed primarily to decrease the time needed to align millions of mouse genomic reads and expressed sequence tags against the human genome sequence. The alignment tools of the time were not capable of performing these operations in a manner that would allow a regular update of the human genome assembly. Compared to pre-existing tools, BLAT was ~500 times faster with performing mRNA/DNA alignments and ~50 times faster with protein/protein alignments.[1]
| Developer(s) | Jim Kent, UCSC |
|---|---|
| Repository | |
| Type | Bioinformatics tool |
| License | free for noncommercial use, commercial use, source available |
| Website | genome |
Overview
BLAT is one of multiple algorithms developed for the analysis and comparison of biological sequences such as DNA, RNA and proteins, with a primary goal of inferring homology in order to discover biological function of genomic sequences.[2] It is not guaranteed to find the mathematically optimal alignment between two sequences like the classic Needleman-Wunsch[3] and Smith-Waterman[4] dynamic programming algorithms do; rather, it first attempts to rapidly detect short sequences which are more likely to be homologous, and then it aligns and further extends the homologous regions. It is similar to the heuristic BLAST[5][6] family of algorithms, but each tool has tried to deal with the problem of aligning biological sequences in a timely and efficient manner by attempting different algorithmic techniques.[2][7]
Uses of BLAT
BLAT can be used to align DNA sequences as well as protein and translated nucleotide (mRNA or DNA) sequences. It is designed to work best on sequences with great similarity. The DNA search is most effective for primates and the protein search is effective for land vertebrates.[1][8] In addition, protein or translated sequence queries are more effective for identifying distant matches and for cross-species analysis than DNA sequence queries.[9] Typical uses of BLAT include the following:
- Alignment of multiple mRNA sequences onto a genome assembly in order to infer their genomic coordinates;[10]
- Alignment of a protein or mRNA sequence from one species onto a sequence database from another species to determine homology. Provided the two species are not too divergent, cross-species alignment is generally effective with BLAT. This is possible because BLAT does not require perfect matches, but rather accepts mismatches in alignments;[11]
- BLAT can be used for alignments of two protein sequences. However, it is not the tool of choice for these types of alignments. BLASTP, the Standard Protein BLAST tool, is more efficient at protein-protein alignments;[1]
- Determination of the distribution of exonic and intronic regions of a gene;[9][10]
- Detection of gene family members of a specific gene query;[9][10]
- Display of the protein-coding sequence of a specific gene.[9][10]
BLAT is designed to find matches between sequences of length at least 40 bases that share ≥95% nucleotide identity or ≥80% translated protein identity.[9][10]
Process
BLAT is used to find regions in a target genomic database which are similar to a query sequence under examination. The general algorithmic process followed by BLAT is similar to BLAST's in that it first searches for short segments in the database and query sequences which have a certain number of matching elements. These alignment seeds are then extended in both directions of the sequences in order to form high-scoring pairs.[12] However, BLAT uses a different indexing approach from BLAST, which allows it to rapidly scan very large genomic and protein databases for similarities to a query sequence. It does this by keeping an indexed list (hash table) of the target database in memory, which significantly reduces the time required for the comparison of the query sequences with the target database. This index is built by taking the coordinates of all the non-overlapping k-mers (words with k letters) in the target database, except for highly repeated k-mers. BLAT then builds a list of all overlapping k-mers from the query sequence and searches for these in the target database, building up a list of hits where there are matches between the sequences[1] (Figure 1 illustrates this process).
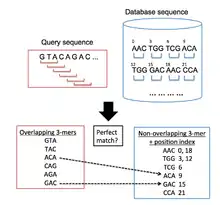
Search stage
There are three different strategies used in order to search for candidate homologous regions:
- The first method requires single perfect matches between the query and database sequences i.e. the two k-mer words are exactly the same. This approach is not considered the most practical. This is because a small k-mer size is necessary in order to achieve high levels of sensitivity, but this increases the number of false positive hits, thus increasing the amount of time spent in the alignment stage of the algorithm.[1]
- The second method allows at least one mismatch between the two k-mer words. This decreases the amount of false positives, allowing larger k-mer sizes which are less computationally expensive to handle than those produced from the previous method. This method is very effective in identifying small homologous regions.[1]
- The third method requires multiple perfect matches which are in close proximity to each other. As Kent shows,[1] this is a very effective technique capable of taking into consideration small insertions and deletions within the homologous regions.
When aligning nucleotides, BLAT uses the third method requiring two perfect word matches of size 11 (11-mers). When aligning proteins, the BLAT version determines the search methodology used: when the client/server version is used, BLAT searches for three perfect 4-mer matches; when the stand-alone version is used, BLAT searches for a single perfect 5-mer between the query and database sequences.[1]
BLAT vs. BLAST
Some of the differences between BLAT and BLAST are outlined below:
- BLAT indexes the genome/protein database, retains the index in memory, and then scans the query sequence for matches. BLAST, on the other hand, builds an index of the query sequences and searches through the database for matches.[1] A BLAST variant called MegaBLAST indexes 4 databases to speed up alignments.[9]
- BLAT can extend on multiple perfect and near-perfect matches (default is 2 perfect matches of length 11 for nucleotide searches and 3 perfect matches of length 4 for protein searches), while BLAST extends only when one or two matches occur close together.[1][9]
- BLAT connects each homologous area between two sequences into a single larger alignment, in contrast to BLAST which returns each homologous area as a separate local alignment. The result of BLAST is a list of exons with each alignment extending just past the end of the exon. BLAT, however, correctly places each base of the mRNA onto the genome, using each base only once and can be used to identify intron-exon boundaries (i.e. splice sites).[1][13]
- BLAT is less sensitive than BLAST.[2]
Program usage
BLAT can be used either as a web-based server-client program or as a stand-alone program.[9]
Server-client
The web-based application of BLAT can be accessed from the UCSC Genome Bioinformatics Site.[8] Building the index is a relatively slow procedure. Therefore, each genome assembly used by the web-based BLAT is associated with a BLAT server, in order to have a pre-computed index available for alignments. These web-based BLAT servers keep the index in memory for users to input their query sequences.[11]
Once the query sequence is uploaded/pasted into the search field, the user can select various parameters such as which species' genome to target (there are currently over 50 species available) and the assembly version of that genome (for example, the human genome has four assemblies to select from), the query type (i.e. whether the sequence relates to DNA, protein etc.) and output settings (i.e. how to sort and visualise the output). The user can then run the search by either submitting the query or using the BLAT "I'm feeling lucky" search.[8]
Bhagwat et al.[9] provide step by step protocols for how to use BLAT to:
- Map an mRNA/cDNA sequence to a genomic sequence;
- Map a protein sequence to the genome;
- Perform homology searches.
Input
BLAT can handle long database sequences, however, it is more effective with short query sequences than long query sequences. Kent[1] recommends a maximum query length of 200,000 bases. The UCSC browser limits query sequences to less than 25,000 letters (i.e. nucleotides) for DNA searches and less than 10,000 letters (i.e. amino acids) for protein and translated sequence searches.[8]
The BLAT Search Genome available on the UCSC website accepts query sequences as text (cut and pasted into the query box) or uploaded as text files. The BLAT Search Genome can accept multiple sequences of the same type at once, up to a maximum of 25. For multiple sequences, the total number of nucleotides must not exceed 50,000 for DNA searches or 25,000 letters for protein or translated sequence searches. An example of searching a target database with a DNA query sequence is shown in Figure 2.
Output
A BLAT search returns a list of results that are ordered in decreasing order based on the score. The following information is returned: the score of the alignment, the region of query sequence that matches to the database sequence, the size of the query sequence, the level of identity as a percentage of the alignment and the chromosome and position that the query sequence maps to.[9] Bhagwat et al.[9] describe how the BLAT "Score" and "Identity" measures are calculated.
For each search result, the user is provided with a link to the UCSC Genome Browser so they can visualise the alignment on the chromosome. This a major benefit of the web-based BLAT over the stand-alone BLAT. The user is able to obtain biological information associated with the alignment, such as information about the gene to which the query may match.[9] The user is also provided with a link to view the alignment of the query sequence with the genome assembly. The matches between the query and genome assembly are blue and the boundaries of the alignments are lighter in colour. These exon boundaries indicate splice sites.[8][9] The "I'm feeling lucky" search result returns the highest scoring alignment for the first query sequence based on the output sort option selected by the user.[8]
License
Both the source and precompiled binaries of BLAT are freely available for academic and personal use. Commercial license of stand-alone BLAT is distributed by Kent Informatics, Inc.
See also
- BLAST Basic Local Alignment Search Tool
- Sequence alignment software
References
- Kent, W James (2002). "BLAT--the BLAST-like alignment tool". Genome Research. 12 (4): 656–664. doi:10.1101/gr.229202. PMC 187518. PMID 11932250.
- Imelfort, Michael (2009). Edwards, D; Stajich, J; Hansen, D (eds.). Bioinformatics: Tools and Applications. New York: Springer. pp. 19–20. ISBN 978-0-387-92737-4.
- Needleman, SB; Wunsch, CD (1970). "A general method applicable to the search for similarities in the amino acid sequence of two proteins". Journal of Molecular Biology. 48 (3): 443–53. doi:10.1016/0022-2836(70)90057-4. PMID 5420325.
- Smith, TF; Waterman, MS (1981). "Identification of common molecular subsequences". Journal of Molecular Biology. 147 (1): 195–7. CiteSeerX 10.1.1.63.2897. doi:10.1016/0022-2836(81)90087-5. PMID 7265238.
- Altschul, SF; Gish, W; Miller, W; Myers, EW; Lipman, DJ (1990). "Basic local alignment search tool". Journal of Molecular Biology. 215 (3): 403–10. doi:10.1016/S0022-2836(05)80360-2. PMID 2231712. S2CID 14441902.
- Altschul, SF; Madden, TL; Schäffer, AA; Zhang, J; Zhang, Z; Miller, W; Lipman, DJ (1997). "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs". Nucleic Acids Research. 25 (17): 3389–402. doi:10.1093/nar/25.17.3389. PMC 146917. PMID 9254694.
- Baxevanis, Andreas D.; Ouellette, B.F. Francis (2001). Bioinformatics: A Practical Guide to the Analysis of Genes and Proteins (2nd ed.). New York: Wiley-Interscience. pp. 187–214. ISBN 978-0-471-22392-4.
- UCSC Genome Bioinformatics Site
- Bhagwat, Medha; Young, Lynn; Robison, Rex R (March 2012). Using BLAT to find sequence similarity in closely related genomes. pp. 10.8.1–10.8.24. doi:10.1002/0471250953.bi1008s37. ISBN 978-0-471-25095-1. PMC 4101998. PMID 22389010.
{{cite book}}:|journal=ignored (help) - Ye, Shui Qing (2008). Bioinformatics: A Practical Approach. London: Chapman & Hall. pp. 11–12. ISBN 978-1-58488-810-9.
- Kuhn, RM; Haussler, D; Kent, WJ (2013). "The UCSC genome browser and associated tools". Briefings in Bioinformatics. 14 (2): 144–61. doi:10.1093/bib/bbs038. PMC 3603215. PMID 22908213.
- Lobo, Ingrid. "Basic Local Alignment Search Tool (BLAST)". Nature Education. Retrieved 15 October 2013.
- Pevsner, J (2009). Bioinformatics and Functional Genomics. New Jersey: John Wiley & Sons, Inc. pp. 166–167. ISBN 978-0-470-08585-1.
- "NCBI – GenBank: AACZ03015565.1". Retrieved 12 October 2013.