Burkitt lymphoma
Burkitt lymphoma is a cancer of the lymphatic system, particularly B lymphocytes found in the germinal center. It is named after Denis Parsons Burkitt, the Irish surgeon who first described the disease in 1958 while working in equatorial Africa.[1][2] The overall cure rate for Burkitt lymphoma in developed countries is about 90%, and it is worse in low-income countries. Burkitt lymphoma is uncommon in adults, in whom it has a worse prognosis.[3]
| Burkitt lymphoma | |
|---|---|
| Other names | Burkitt's tumor, Burkitt's lymphoma, malignant lymphoma Burkitt's type |
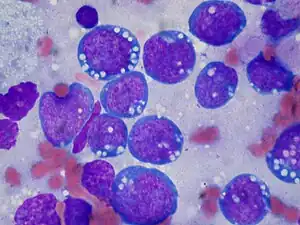 | |
| Burkitt lymphoma, touch prep, Wright stain | |
| Specialty | Hematology and oncology |
Classification
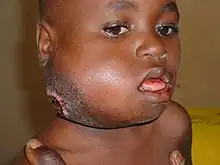
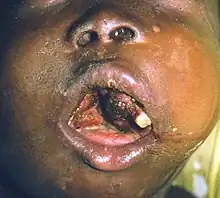
Burkitt lymphoma can be divided into three main clinical variants: the endemic, the sporadic, and the immunodeficiency-associated variants.[3] By morphology (i.e., microscopic appearance), immunophenotype, and genetics, the variants of Burkitt lymphoma are alike.[3]
- The endemic variant (also called "African variant") most commonly occurs in children living in regions where malaria is endemic (such as equatorial Africa, Brazil, and Papua New Guinea).[4] Epstein–Barr virus (EBV) infection is found in nearly all patients.[3] Chronic malaria is believed to reduce resistance to EBV, allowing EBV infection to occur. The disease characteristically involves the jaw or other facial bone, abdomen, cecum, distal ileum ovaries, kidney, or breast.[4][5] Less than 10% of cases present with central nervous system involvement, which usually manifest as cranial nerve palsies or spinal cord compression.[5]
- The sporadic type of Burkitt lymphoma (also known as "non-African") is the most common variant found in places where malaria is not endemic such as North America and parts of Europe.[3] The median onset is 10 years of age, but there are also peaks at ages 40 and 75 years old.[5] Males are 3-4 times more likely to be affected as compared to females.[5] The tumor cells have a similar appearance to the those of the classical endemic Burkitt lymphoma. Sporadic type Burkitt lymphomas are less commonly associated with the EBV virus as compared to the endemic variant; with 20-30% of cases being attributed to EBV (most commonly in adults older than 50).[5] The jaw is less commonly involved in this variant.[3] The abdominal region is the most common site of involvement, often at the right lower quadrant.[3][5] The bone marrow is involved in 30-35% of cases and the central nervous system is involved in approximately 20% of cases (usually affecting the meninges, or the covering of the brain and spinal cord).[5]
- Immunodeficiency-associated Burkitt lymphoma is usually associated with HIV infection, but can also occur in the setting of post-transplant patients.[6][3] The median age of onset of the immunodeficiency-associated variant is between 40-45 years of age and it is equally prevalent in males and females.[5] It makes up 40% of the HIV associated lymphomas, and it usually occurs in those with normal CD4+ T cell counts.[5] EBV is detected in 25-40% of cases and common areas of involvement include GI tract, lymph nodes and bone marrow with 20-30% of affected people having central nervous system involvement.[5]
Burkitt lymphoma is commonly associated with the infection of B cell lymphocytes with the EBV and in these cases is considered to be one form of the Epstein–Barr virus-associated lymphoproliferative diseases.[7] The endemic variant of Burkitt lymphoma is in almost all cases associated with EBV infection.[8] The fact that some Burkitt lymphoma cases do not involve EBV allows that many cases of the disease are not caused and/or promoted by EBV, i.e. the virus may be an innocent passenger virus in these cases. However, the almost ubiquitous presence of the virus in the endemic variant of Burkitt lymphoma suggests that it contributes to the development and/or progression of this variant.[9] The mutational landscape in Burkitt lymphoma has recently been found to differ between tumors with and without EBV infection, further strengthening the role of the virus in disease origin.[10]
Pathophysiology
Genetics
All types of Burkitt lymphoma are characterized by dysregulation of the c-myc gene by one of three chromosomal translocations which place the myc gene under the control of an immunoglobulin gene enhancer.[11] The MYC gene is found at 8q24.
- The most common variant is t(8;14)(q24;q32), which accounts for about 70 to 80% of cases of cases.[5] This involves c-myc oncogene translocation from chromosome 8 to the Ig heavy chain region of chromosome 14. A variant of this, a three-way translocation, t(8;14;18), has also been identified.[12]
- Another variant is t(2;8)(p12;q24).[13] This involves the myc oncogene being translocated from chromosome 8 to the Ig kappa locus on chromosome 2. This type of translocation is seen in 15% of cases of Burkitt lymphoma.
- A rare variant is t(8;22)(q24;q11).[13] This type involves myc oncogene translocation from chromosome 8 to the Ig lambda locus on chromosome 22. This type of translocation is involved in about 5% of cases of Burkitt lympohoma.
The c-myc gene found on chromosome 8 is part of the Myc family of genes that serve as regulators of cellular transcription and is associated with Burkitt lymphoma.[14][15] Expression of the c-myc gene results in the synthesis of transcriptional factors that increase the expression of other genes involved in aerobic glycolysis.[14] Ultimately, an increase in aerobic glycolysis plays a role in providing the necessary energy for cellular growth to occur.[14] The translocation of the c-myc gene to the IGH, IGK, or IGL region moves the gene to a location in the genome near immunoglobulin enhancers which increases the expression of the c-myc gene.[14] Overall, this translocation leads to increased cellular proliferation that is found in Burkitt lymphoma.[14] Point mutations can also be present in the translocated c-myc gene resulting in the expressed c-myc protein being overactive.[14]
Bcl-2 translocations, which are frequently seen in follicular lymphomas and other B-cell Non-Hodgkin Lymphomas, do not occur in Burkitt lymphomas.[5]
One of the above described translocations of Myc is seen in 90% of cases of Burkitt lymphoma, but these oncogenic translocations are not usually sufficient to cause lymphoma; other mutations must also be present.[5] These additional mutations include mutations of the tumor suppressor TP53, which interacts with the tumor suppressor p53 (which usually causes apoptosis in B cells carrying the disordered MYC oncoprotein). But with TP53 and p53 mutated, apoptosis is blocked, and the oncogenic B-cells are allowed to proliferate unchecked.[5] The tumor suppressors ARF and USP7 are also frequently mutated in Burkitt lymphoma leading to MDM2 inhibition of the tumor suppressor p53 which then leads to enhanced oncogenesis.[5] SIN3A, a regulator of MYC, that acts to inhibit MYC by deacetylating it, is often inactivated in Burkitt lymphoma.[5] Also, sequential mutations of the RNA helicases (involved in RNA synthesis) DDX3X (found on the x chromosome) and DDX3Y (found on the y chromosome) lead to MYC oncogenesis in Burkitt lymphoma.[16] Early in the pathogenesis process, DDX3X mutations limit translation (protein synthesis) allowing lymphoma cells to escape MYC induced proteotoxic stress and apoptosis, then later, DDX3Y mutations restore high level protein synthesis (by producing the translational machinery) and leading to increased proliferation of tumor cells.[16] These sequential DDX3X and DDX3Y mutations are thought to partially explain why Burkitt lymphoma is more common in males as the DDX3Y RNA helicase is only found on the Y chromosome.[5] Mutations affecting the transcription factor TCF3 and its negative regulator ID3 are found in about 70% of cases of Burkitt lymphoma.[5] These mutations prevent ID3 from binding to and inhibiting TCF3; thus the hyperactive TCF3 then activates B cell receptors which activate PI3K and mTOR, as well as Ig heavy and light chain genes, which contribute to oncogenesis.[14][5] TCF3 and ID3 mutations lead to continuously active B-cell receptors, explaining the high level of proliferation seen in Burkitt lymphoma.[5] The cell cycle regulators Cyclin D3 and p16 may also be activated and deactivated respectively in Burkitt lymphoma; leading to massive tumor cell proliferation.[5]
Some epigenetic mechanisms have been found to play a role in the pathogenesis of Burkitt lymphoma. FBXO11 is a chromatin regulator. By activating ubiquitin ligase, FBXO11 causes ubiquitination of BCL6 which causes it to be targeted for proteasome degradation.[5] BCL6 normally helps B cells mature in the germinal center and produce antibodies specific to encountered antigens. In Burkitt lymphoma, FBXO11 is deactivated, leading to increased BCL6 activation which then leads to increased proliferation and decreased maturation of germinal center B-cells, thus promoting lymphomagenesis.[5]
EBV associated Burkitts has increased expressional activity of activation-induced cytidine deaminase, which is a mutator, this leads to EBV associated Burkitt lymphomas having more mutations than non-EBV types. Non-EBV subtypes of Burkitt lymphoma more commonly have dysregulation of cyclin D3 and mutated, inactivated p53.[5]
Virology
The complete role of EBV in the pathogenesis of endemic Burkitt lymphoma is not completely elucidated, but it has been shown to cause DNA damage, dysfunction of telomeres, and genome instability.[17][15] B cell infection with EBV is latent, and the virus does not undergo replication.[17] These latently infected B cells can then go on to produce proteins that function to promote cellular growth through modification of normal signaling pathways.[17] EBV promotes the development of malignant B cells via proteins that limit apoptosis in cells that had the c-myc translocation.[15] Apoptosis is limited by EBV through various means such as the EBNA-1 protein, BHF1 protein, EBER transcripts, vIL-10 gene, BZLF1, and LMP1.[15] Malaria has been found to cause genomic instability in endemic Burkitt lymphoma.[18] Malaria can lead to the reactivation of latent EBV and also MYC translocations via activation of the toll-like receptor 9.[15] Malaria also promotes B-cell proliferation by altering the regular immune response.[17] The immune system targets antigens (e.g., EBNA2 and LMP-1) and eliminates most B cells infected with EBV.[15] Downregulation of antigens targeted by the immune system leads to the development of persistent B cells.[15] These B cells can then undergo further mutations (e.g., c-myc translocation) that promote cancer development.[15]
Immunology
Burkitt Lymphoma cells express HLA class I molecules normally, as well as some HLA class II complexes; however, CD4+ T cells are not properly activated. Burkitt lymphoma cells only express EBNA 1, a poorly antigenic EBV-associated antigen, that is able to escape HLA class I presentation, thus evades an immune response. EBNA 1 can be presented via HLA class II molecules, however HLA Class II pathway is unable to activate the CD4+ T cells.[19]
Diagnosis
Malignant B cell characteristics
Normal B cells of a germinal center possess rearranged immunoglobulin heavy and light chain genes, and each isolated B cell possesses a unique IgH gene rearrangement. Since Burkitt lymphoma and other B-cell lymphomas are a clonal proliferative process, all tumor cells from one patient are supposed to possess identical IgH genes. When the DNA of tumor cells is analyzed using electrophoresis, a clonal band can be demonstrated, since identical IgH genes will move to the same position. On the contrary, when a normal or reactive lymph node is analyzed using the same technique, a smear rather than a distinct band will be seen. This technique is useful since sometimes benign reactive processes (e.g. infectious mononucleosis) and malignant lymphoma can be difficult to distinguish.
Microscopy
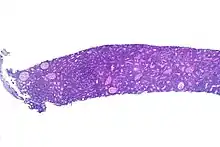
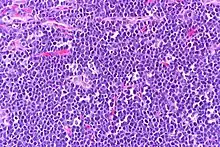
The tumor consists of sheets of a monotonous (i.e., similar in size and morphology) population of medium-sized lymphoid cells with high proliferative and apoptotic activity. The "starry sky" appearance seen[20] under low power is due to scattered tingible body-laden macrophages (macrophages containing dead apoptotic tumor cells). The old descriptive term of "small non-cleaved cell" is misleading. The tumor cells are mostly medium in size (i.e., tumor nuclei size similar to that of histiocytes or endothelial cells). "Small non-cleaved cells" are compared to "large non-cleaved cells" of normal germinal center lymphocytes. Tumor cells possess small amounts of basophilic cytoplasm with three to four small nucleoli. The cellular outline usually appears squared off.
Immunohistochemistry
The tumor cells in Burkitt lymphoma generally strongly express markers of B cell differentiation (CD20, CD22, CD19), as well as CD10 and BCL6. The tumor cells are generally negative for BCL2 and TdT. The high mitotic activity of Burkitt lymphoma is confirmed by nearly 100% of the cells staining positive for Ki67.[21]
Treatment
In general, the first line of treatment for Burkitt lymphoma is chemotherapy. A few of these regimens are: the GMALL-B-ALL/NHL2002 protocol, the modified Magrath regimen (R-CODOX-M/IVAC).[22] COPADM,[23] hyper-CVAD,[24] and the Cancer and Leukemia Group B (CALGB) 8811 regimen;[24] these can be associated with rituximab.[24][25] In older patients, treatment may be dose-adjusted EPOCH with rituximab.[26]
The effects of the chemotherapy, as with all cancers, depend on the time of diagnosis. With faster-growing cancers, such as Burkitt, the cancer actually responds faster than with slower-growing cancers. This rapid response to chemotherapy can be hazardous to the patient, as a phenomenon called "tumor lysis syndrome" could occur. Close monitoring of the patient and adequate hydration is essential during the process. Since Burkitt lymphoma has high propensity to spread to the central nervous system (lymphomatous meningitis), intrathecal chemotherapy with methotrexate and/or ARA-C and/or prednisolone is given along with systemic chemotherapy.
Other treatments for Burkitt lymphoma include immunotherapy, bone marrow transplants, stem cell transplant, surgery to remove the tumor, and radiotherapy.
Prognosis
The prognosis for Burkitt lymphoma is superior in children and increased age is a negative prognostic indicator.[28][15] The overall cure rate for sporadic Burkitt lymphoma in developed countries is about 90%.[3] In adults, Burkitt lymphoma is uncommon and has poor outcomes.[3] Unsatisfactory initial response to the treatment of Burkitt lymphoma with cyclophosphamide, vincristine, and prednisolone indicates a negative prognosis.[15] Furthermore, failed initial treatment and return of Burkitt lymphoma after a six-month stint of time serve as a poor prognostic indicator.[15] The adequate utilization of therapeutic drugs during initial treatment limits additional treatment options following the return of the disease.[15] Notably, in areas of the world where the initial treatment of Burkitt lymphoma is inadequate further treatment options may remain for cases when the disease returns.[15] Laboratory studies such as lactate dehydrogenase (LDH), CD4 count, and other cytogenetic studies are also prognostic indicators.[15] Unsatisfactory outcomes have been associated with an LDH that is found to be two times above the upper limit of normal.[15] Specifically, there is a poor prognosis associated with a CD4 count that is decreased in the immunodeficiency-associated variant of Burkitt lymphoma.[15] Genetic mutations extending beyond the previously described MYC translocation may also serve as negative prognostic indicators.[15] Some notable genetic findings that may be associated with poor outcomes include: 13q deletion, 7q gain, ID3 and CCND3 double-hit mutations, and 18q21 CN-LOH mutations.[15] The prognosis for Burkitt lymphoma can be better determined following staging utilizing imaging modalities such as positron emission tomography and computed tomography scans where tumor burden and invasion of the central nervous system have been found to indicate a poor prognosis.[28][15]
Epidemiology
As a non-Hodgkin lymphoma (NHL), Burkitt lymphoma makes up 1-5% of cases, and it is more common in males than females with a 3–4 to 1 ratio.[15] The endemic variant mainly impacts areas with an increased prevalence of malaria and EBV in Africa and Papua New Guinea.[15][29] For children less than 18 years of age from equatorial Africa, the annual incidence of Burkitt lymphoma is 4–5/100,000.[29] Additionally, in equatorial Africa, 50% of tumors that are diagnosed during childhood as well as 90% of lymphoma cases can be attributed to Burkitt lymphoma.[29] The peak incidence for endemic Burkitt lymphoma is from ages 4 to 7 with an average age of 6 years.[15][29] The sporadic variant with an annual incidence 2-3/million is more commonly found in North America and Europe comprising 1-2% of adult lymphomas and 30–40% of NHL cases.[15][29] This variant is 3.5 times more commonly found in males compared to females and it is more frequent in younger individuals.[29] The sporadic variant has a peak incidence at 11 years of age in children, and diagnosis typically occurs from 3–12 years of age on average.[15][29] For adults, 45 years was the median age that the sporadic Burkitt lymphoma was diagnosed.[15] The immunodeficiency-associated variant predominantly impacts the HIV-infected population.[29] For those in the United States and with AIDS, the incidence of this variant was found to be 22/100,000 person-years.[15][29] There is also an increased risk of developing this variant of Burkitt lymphoma for individuals that have received an organ transplant after 4–5 years.[29]
EBV infection is associated with Burkitt lymphoma.[30] EBV is found in virtually all instances of endemic Burkitt lymphoma.[29] The sporadic variant of Burkitt lymphoma is associated with EBV in only 10–20% of cases.[29] Approximately 30% of immunodeficiency-associated Burkitt lymphoma cases were associated with EBV.[31]
Research
Gene targets
Unique genetic alterations promote cell survival in Burkitt lymphoma, distinct from other types of lymphoma.[32] These TCF3 and ID3 gene mutations in Burkitt correspond to a cell survival pathway that may be found to be amenable to targeted therapy.[33]
References
- synd/2511 at Who Named It?
- Burkitt D (1958). "A sarcoma involving the jaws in African children". The British Journal of Surgery. 46 (197): 218–23. doi:10.1002/bjs.18004619704. PMID 13628987. S2CID 46452308.
- Molyneux E, Rochford R, Griffin B, Newton R, Jackson G, Menon G, Harrison C, Israels T, Bailey S (April 2012). "Burkitt's lymphoma" (PDF). The Lancet. 379 (9822): 1234–1244. doi:10.1016/S0140-6736(11)61177-X. PMID 22333947. S2CID 39960470.
- Dojcinov, SD; Fend, F; Quintanilla-Martinez, L (7 March 2018). "EBV-Positive Lymphoproliferations of B- T- and NK-Cell Derivation in Non-Immunocompromised Hosts". Pathogens (Basel, Switzerland). 7 (1): 28. doi:10.3390/pathogens7010028. PMC 5874754. PMID 29518976.
- Roschewski, Mark; Staudt, Louis M.; Wilson, Wyndham H. (22 September 2022). "Burkitt's Lymphoma". New England Journal of Medicine. 387 (12): 1111–1122. doi:10.1056/NEJMra2025746. PMID 36129999. S2CID 252437964.
- Bellan C, Lazzi S, De Falco G, Nyongo A, Giordano A, Leoncini L (March 2003). "Burkitt's lymphoma: new insights into molecular pathogenesis". J. Clin. Pathol. 56 (3): 188–92. doi:10.1136/jcp.56.3.188. PMC 1769902. PMID 12610094.
- Vockerodt M, Yap LF, Shannon-Lowe C, Curley H, Wei W, Vrzalikova K, Murray PG (January 2015). "The Epstein-Barr virus and the pathogenesis of lymphoma". The Journal of Pathology. 235 (2): 312–22. doi:10.1002/path.4459. PMID 25294567. S2CID 22313509.
- Casulo C, Friedberg J (September 2015). "Treating Burkitt Lymphoma in Adults". Current Hematologic Malignancy Reports. 10 (3): 266–71. doi:10.1007/s11899-015-0263-4. PMID 26013028. S2CID 21258747.
- Rezk SA, Zhao X, Weiss LM (September 2018). "Epstein-Barr virus (EBV)-associated lymphoid proliferations, a 2018 update". Human Pathology. 79: 18–41. doi:10.1016/j.humpath.2018.05.020. PMID 29885408. S2CID 47010934.
- Grande, Bruno M.; Gerhard, Daniela S.; Jiang, Aixiang; Griner, Nicholas B.; Abramson, Jeremy S.; Alexander, Thomas B.; Allen, Hilary; Ayers, Leona W.; Bethony, Jeffrey M.; Bhatia, Kishor; Bowen, Jay; Casper, Corey; Choi, John Kim; Culibrk, Luka; Davidsen, Tanja M.; Dyer, Maureen A.; Gastier-Foster, Julie M.; Gesuwan, Patee; Greiner, Timothy C.; Gross, Thomas G.; Hanf, Benjamin; Harris, Nancy Lee; He, Yiwen; Irvin, John D.; Jaffe, Elaine S.; Jones, Steven J. M.; Kerchan, Patrick; Knoetze, Nicole; Leal, Fabio E.; Lichtenberg, Tara M.; Ma, Yussanne; Martin, Jean Paul; Martin, Marie-Reine; Mbulaiteye, Sam M.; Mullighan, Charles G.; Mungall, Andrew J.; Namirembe, Constance; Novik, Karen; Noy, Ariela; Ogwang, Martin D.; Omoding, Abraham; Orem, Jackson; Reynolds, Steven J.; Rushton, Christopher K.; Sandlund, John T.; Schmitz, Roland; Taylor, Cynthia; Wilson, Wyndham H.; Wright, George W.; Zhao, Eric Y.; Marra, Marco A.; Morin, Ryan D.; Staudt, Louis M. (21 March 2019). "Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma". Blood. 133 (12): 1313–1324. doi:10.1182/blood-2018-09-871418. PMC 6428665. PMID 30617194.
- Hoffman, Ronald (2009). Hematology : basic principles and practice (PDF) (5th ed.). Philadelphia, PA: Churchill Livingstone/Elsevier. pp. 1304–1305. ISBN 978-0-443-06715-0.
- Liu D, Shimonov J, Primanneni S, Lai Y, Ahmed T, Seiter K (2007). "t(8;14;18): a 3-way chromosome translocation in two patients with Burkitt's lymphoma/leukemia". Mol. Cancer. 6 (1): 35. doi:10.1186/1476-4598-6-35. PMC 1904237. PMID 17547754.
- Smardova J, Grochova D, Fabian P, et al. (October 2008). "An unusual p53 mutation detected in Burkitt's lymphoma: 30 bp duplication". Oncol. Rep. 20 (4): 773–8. doi:10.3892/or_00000073. PMID 18813817.
- Robbins & Cotran pathologic basis of disease. Vinay Kumar, Abul K. Abbas, Jon C. Aster (10th ed.). Philadelphia, PA. 2021. pp. 583–633. ISBN 978-0-323-53113-9. OCLC 1191840836.
{{cite book}}: CS1 maint: location missing publisher (link) CS1 maint: others (link) - Graham, Brittney S.; Lynch, David T. (2022), "Burkitt Lymphoma", StatPearls, Treasure Island (FL): StatPearls Publishing, PMID 30844175, retrieved 2022-01-20
- Gong, Chun; Krupka, Joanna A.; Gao, Jie; Grigoropoulos, Nicholas F.; Giotopoulos, George; Asby, Ryan; Screen, Michael; Usheva, Zelvera; Cucco, Francesco; Barrans, Sharon; Painter, Daniel; Zaini, Nurmahirah Binte Mohammed; Haupl, Björn; Bornelöv, Susanne; Ruiz De Los Mozos, Igor; Meng, Wei; Zhou, Peixun; Blain, Alex E.; Forde, Sorcha; Matthews, Jamie; Khim Tan, Michelle Guet; Burke, G.A. Amos; Sze, Siu Kwan; Beer, Philip; Burton, Cathy; Campbell, Peter; Rand, Vikki; Turner, Suzanne D.; Ule, Jernej; Roman, Eve; Tooze, Reuben; Oellerich, Thomas; Huntly, Brian J.; Turner, Martin; Du, Ming-Qing; Samarajiwa, Shamith A.; Hodson, Daniel J. (October 2021). "Sequential inverse dysregulation of the RNA helicases DDX3X and DDX3Y facilitates MYC-driven lymphomagenesis". Molecular Cell. 81 (19): 4059–4075.e11. doi:10.1016/j.molcel.2021.07.041. PMID 34437837. S2CID 237327258.
- Robbins & Cotran pathologic basis of disease. Vinay Kumar, Abul K. Abbas, Jon C. Aster, Ramzi S. Cotran, Stanley L. Robbins (10th ed.). Philadelphia, Pa. 2021. pp. 267–338. ISBN 978-0-323-60995-1. OCLC 1161987164.
{{cite book}}: CS1 maint: location missing publisher (link) CS1 maint: others (link) - Hematology : basic principles and practice. Ronald Hoffman, Edward J., Jr. Benz, Leslie E. Silberstein, Helen Heslop, Jeffrey I. Weitz, John Anastasi (7th ed.). Philadelphia, PA. 2018. pp. 1187–1203. ISBN 978-0-323-50939-8. OCLC 1001961209.
{{cite book}}: CS1 maint: location missing publisher (link) CS1 maint: others (link) - God, Jason; Haque, Azizul (2010). "Burkitt Lymphoma: Pathogenesis and Immune Evasion". Journal of Oncology. 2010: 1–14. doi:10.1155/2010/516047. PMC 2952908. PMID 20953370.
- Fujita S, Buziba N, Kumatori A, Senba M, Yamaguchi A, Toriyama K (May 2004). "Early stage of Epstein–Barr virus lytic infection leading to the "starry sky" pattern formation in endemic Burkitt's lymphoma". Arch. Pathol. Lab. Med. 128 (5): 549–52. doi:10.5858/2004-128-549-ESOEVL. PMID 15086279.
- Steven H Swerdlow (2008). WHO classification of tumours of haematopoietic and lymphoid tissues. World Health Organization classification of tumours. Lyon, France : International Agency for Research on Cancer. ISBN 978-92-832-2431-0.
- Barnes, J.A.; LaCasce2, A.S; Feng, Y.; et al. (2011). "Evaluation of the addition of rituximab to CODOX-M/ IVAC for Burkitt's lymphoma: a retrospective analysis". Annals of Oncology. 22 (8): 1859–64. doi:10.1093/annonc/mdq677. PMID 21339382.
- Miles, Rodney R.; Arnold, Staci; Cairo, Mitchell S. (2012). "Risk factors and treatment of childhood and adolescent Burkitt lymphoma/leukaemia". British Journal of Haematology. 156 (6): 730–743. doi:10.1111/j.1365-2141.2011.09024.x. PMID 22260323. S2CID 6418151.
- "Burkitt Lymphoma and Burkitt-like Lymphoma: Practice Essentials, Background, Etiology and Pathophysiology". 29 June 2017. Retrieved 19 March 2018 – via eMedicine.
{{cite journal}}: Cite journal requires|journal=(help) - "BHS Guidelines for the treatment of Burkitt's lymphoma" (PDF). Bhs.be. Retrieved 17 March 2022.
- Wyndham H. Wilson; Kieron Dunleavy; Stefania Pittaluga; Upendra Hegde; Nicole Grant; Seth M. Steinberg; Mark Raffeld; Martin Gutierrez; Bruce A. Chabner; Louis Staudt; Elaine S. Jaffe; John E. Janik (2008). "Phase II Study of Dose-Adjusted EPOCH-Rituximab in Untreated Diffuse Large B-cell Lymphoma with Analysis of Germinal Center and Post-Germinal Center Biomarkers". Journal of Clinical Oncology. 26 (16): 2717–2724. doi:10.1200/JCO.2007.13.1391. PMC 2409217. PMID 18378569.
- Yustein JT, Dang CV (2007). "Biology and treatment of Burkitt's lymphoma". Curr. Opin. Hematol. 14 (4): 375–81. doi:10.1097/MOH.0b013e3281bccdee. PMID 17534164. S2CID 8778208.
- Hematology : basic principles and practice. Ronald Hoffman, Edward J., Jr. Benz, Leslie E. Silberstein, Helen Heslop, Jeffrey I. Weitz, John Anastasi (7th ed.). Philadelphia, PA. 2018. pp. 1309–1317. ISBN 978-0-323-50939-8. OCLC 1001961209.
{{cite book}}: CS1 maint: location missing publisher (link) CS1 maint: others (link) - Saleh, Khalil; Michot, Jean-Marie; Camara-Clayette, Valérie; Vassetsky, Yegor; Ribrag, Vincent (2020-03-06). "Burkitt and Burkitt-Like Lymphomas: a Systematic Review". Current Oncology Reports. 22 (4): 33. doi:10.1007/s11912-020-0898-8. ISSN 1534-6269. PMID 32144513. S2CID 212420935.
- Pannone, Giuseppe; Zamparese, Rosanna; Pace, Mirella; Pedicillo, Maria; Cagiano, Simona; Somma, Pasquale; Errico, Maria; Donofrio, Vittoria; Franco, Renato; De Chiara, Annarosaria; Aquino, Gabriella; Bucci, Paolo; Bucci, Eduardo; Santoro, Angela; Bufo, Pantaleo (2014). "The role of EBV in the pathogenesis of Burkitt's Lymphoma: an Italian hospital based survey". Infectious Agents and Cancer. 9 (1): 34. doi:10.1186/1750-9378-9-34. ISSN 1750-9378. PMC 4216353. PMID 25364378.
- Navari M, Etebari M, De Falco G, Ambrosio MR, Gibellini D, Leoncini L, Piccaluga PP (2015). "The presence of Epstein-Barr virus significantly impacts the transcriptional profile in immunodeficiency-associated Burkitt lymphoma". Frontiers in Microbiology. 6: 556. doi:10.3389/fmicb.2015.00556. PMC 4462103. PMID 26113842.
- "NIH study shows Burkitt lymphoma is molecularly distinct from other lymphomas". National Cancer Institute. Archived from the original on 2012-08-16. Retrieved 2012-10-19.
- Staudt L; et al. (2012). "Burkitt Lymphoma Pathogenesis and Therapeutic Targets from Structural and Functional Genomics". Nature. 490 (7418): 116–120. Bibcode:2012Natur.490..116S. doi:10.1038/nature11378. PMC 3609867. PMID 22885699.