C5-convertase
C5 convertase is an enzyme belonging to a family of serine proteases that play key role in the innate immunity. It participates in the complement system ending with cell death.
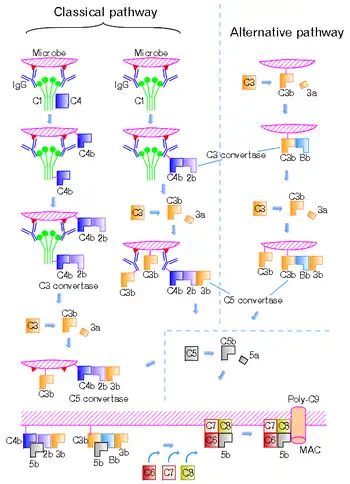

There are four different C5 convertases able to specifically convert the protein C5 to C5a and C5b fragments. Two of the convertases are physiological complement enzymes, associate to the cell-surface and mediate the classical pathway (C4b2b3b, or C4b2a3b depending on source)[1] or the alternative pathway (C3bBbC3b) of complement system.[2][3] Two fluid phase C5 convertases have been described: the classical pathway enzyme, C4b2boxy3b and the cobra venom factor-dependent C5 convertase, CVFBb.
Structure
Cell-bound C3 and C5 convertase differ in their C3b requirement. C3-convertase (C3bBb) need only one molecule of C3b to form, whereas two or more C3b are required for generation of C5 convertase (C3bBb). It means, when C3b is randomly distributed on the surface of a cell, only C3 convertase activity appears after addition of Factors B and D. However, when C3b is distributed in clusters, C3 and C5 convertase activity is generated upon addition of Factors B and D.[3]
The classical pathway C5 convertase is composed of the fragments of complement proteins, C4b, C2a produced by cleavage mediated by C1 complex, and C3b produced by cleavage mediated by the classical pathway C3 convertase (C4bC2a). The formation of the alternative pathway C5 convertase (C3bBbC3b) starts by spontaneous cleavage of C3 protein exposing previously hidden thioester bond. In the presence of pathogen the fragment C3b binds to microbial cell-surface through the newly showed thioester bond. On the other hand, if the infection does not occur, C3b interacts with molecules of water, therefore the protein becomes inactive. However, when C3b undergoes its post-cleavage conformational change, a binding site for a plasma protein called Factor B is also exposed. Factor B then binds to C3b and is cleaved by a plasma serine protease Factor D. The C3bBb complex (= alternative pathway C3 convertase) remains attached to the cell-surface. This complex might interact with another C3b and thus form the alternative pathway C5 convertase.[4] CVFBb is a noncovalent association product of CVF3 and the complement fragment Bb. The catalytic subunits of these multimolecular proteases are C2b and Bb. These subunits belong to atypical serine proteases.[5][6] CVFBb does not require C3 for cleavage of C5, whereas C4b2boxy need native C3 for cleavage of C5 protein. The modified C5 convertase, C4b2boxy3b, contains C2b that is derived from C2 oxidized by iodine.
Function
The target of C5 convertase is complement protein C5. C5 is a two-chain (α, β) plasma glycoprotein (Mr = 196,000). C5 and C3 have similar structure. However, C5 does not appear to contain the internal thiol ester group reported for C3 and C4. C5 has relatively few disulfide bonds. There are three disulfide bonds in C5a, the α-chain has 15 half-Cystines, and the β-chain has only 6 half-Cystines. This comparatively low level of stabilizing disulfide bridges may provide a partial explanation for the irreversible conformational change imparted on C5 after cleavage to C5a and C5b. In addition, the relatively low number of disulfide bonds could account for instability of C5 when exposed to chaotropic agents such as potassium thiocyanate.[2] Electron micrographs of negatively stained C5 indicate that the protein is irregular in shape and contains several lobes.[7]
First of all, C5 has to bind to C3b fragment. The capacity to bind C3b is a stable feature of component C5, as C5b also has this binding capacity. The C5 convertase selectively cleaves an Arginyl-Leucine peptide bond at position 74-75 in the α-chain (Mr = 116,000) of C5. Research has shown that during the classical pathway of the complement system, an inactive A6 allotype of c4 completely stalls the molecules' ability to act as a c5 binding subunit1.[8] This defect in C4A6 activity happens during the C5 binding step to the 4b and c3b complex.[9] α´-chain (Mr, = 105,000) and the activation peptide, C5a, is formed, while β-chain (Mr = 80,000) remains unchanged.[2]
The complement component C5 can be also activated by fluid phase C5 convertase. C5 is activated by CVFBb in the presence of complement component C6 and the C5b6 complex is formed. However, when C6 is added after C5 has been converted to C5b, the C5b6 complex fails to form. Therefore, the activation of C5 results in a transient binding site for C6. Hydrophobic sites are probably exposed upon C5 activation because C5b undergoes aggregation when C5 is converted to C5b in the absence of C6. Interactions between C5 and C6 or C5 and membranes are noncovalent. (In contrast, it is the labile thiol ester that permits covalent attachment between C3 and nucleophilic acceptors.) The proteolytic cleavage of C5 is the only known enzymatic event in assembly of the cytolytic membrane attack complex of complement.[7]
Once bound, C5 is exceptionally efficient in producing hemolysis, requiring less than seven specifically bound molecules per cell for the production of a hemolytic lesion. The extent of formation of the C5 intermediate complex is primarily dependent on the number of molecules of C4, C2 and C3 present on the cells employed for its generation. In these respects, the mode of action of C5 is completely analogous to that of the other components of complement. The C5 step differs, however, in other aspects. The binding of C5 is influenced by C6 and C7, components which are thought to act subsequent to it in the complement sequence. In addition, the hemolytic activity of the isolated C5 intermediate complex is exceedingly labile, having an average half-life at 30 °C of only 9 rain. This characteristic distinguishes the C5 step, along with the C2 step, as potentially rate-limiting in the complement reaction. However, unlike C2, C5 remains firmly cell-bound during the decay process and apparently undergoes an alteration in situ which renders it hemolyticly unreactive. Finally, C5 is unique in that it readily adsorbs in native form to unsensitized erythrocytes. This nonspecifically bound C5 remains firmly attached, although it may be specifically utilized as a source of C5 by an ongoing complement reaction.[1]
Stabilization and regulation
Both enzymes, C4b2b3b and C3bBbC3b, are unstable and undergo decay dissociation with a half-life at 37 °C of approximately 1.5 - 3 min.[1] The properdin stabilizes the alternative pathway C5 convertase of which half-life is at 37 °C 10 - 34 min.[2][3] In contrast, the fluid phase C5 convertase CVFBb is stable (half-life at 37 °C = 7 h).[10] The oxidation of C2 protein stabilizes the C4b2boxy complex.[11] The Factor H–related protein 1 (FHR1) has been identified as a novel inhibitor of the complement pathway. FHR1 blocks C5 convertase activity and interferes with C5b surface deposition and membrane attack complex (MAC) formation. Apparently Factor H and FHR1 control complement activation in a sequential manner. In hemolytic uremic syndrome (HUS), the absence of FHR1 may result in reduced inhibition of terminal complex formation and in reduced protection of endothelial cells upon complement attack.[12]
[13]==References==
- Cooper NR, Müller-Eberhard HJ (1970). "The Reaction Mechanism of Human C5 in Immune Hemolysis". J Exp Med. 132 (4): R775–793. doi:10.1084/jem.132.4.775. PMC 2138854. PMID 5508377.
- DiScipio RG (1982). "The activation of the alternative pathway C3 convertase by human plasma kallikrein". Immunology. 45 (3): R587–595. PMC 1555245. PMID 6916710.
- Medicus RG, Götze O, Müller-Eberhard HJ (1976). "Alternative pathway of complement: Recruitment of precursor properdin by the labile C3/C5 convertase and the potentiation of the pathway". J Exp Med. 144 (4): R1076–1093. doi:10.1084/jem.144.4.1076. PMC 2190426. PMID 978134.
- Abbas AK, Lichtman AH, Pillai S (2010). Cellular and Molecular Immunology (6th ed.). Elsevier. ISBN 978-1-4160-3123-9.
- Kerr MA, Gagnon J (1982). "The purification and properties of the second component of guinea-pig complement". Biochem J. 205 (1): R59–67. doi:10.1042/bj2050059. PMC 1158446. PMID 6922702.
- Christie DL, Gagnon J, Porter RR (1980). "Partial sequence of human complement component Factor B: Novel type of serine protease". Proc. Natl. Acad. Sci. USA. 77 (8): R4923–4927. Bibcode:1980PNAS...77.4923C. doi:10.1073/pnas.77.8.4923. PMC 349961. PMID 6776529.
- DiScipio RG, Smith CA, Müller-Eberhard HJ, Hugli TE (1983). "The Activation of Human Complement Component C5 by a Fluid Phase C5 Convertase". J Biol Chem. 258 (17): R10629–10636. doi:10.1016/S0021-9258(17)44503-0. PMID 6554279.
- Ebanks, R. O.; Jaikaran, A. S.; Carroll, M. C.; Anderson, M. J.; Campbell, R. D.; Isenman, D. E. (1 May 1992). "A single arginine to tryptophan interchange at beta-chain residue 458 of human complement component C4 accounts for the defect in classical pathway C5 convertase activity of allotype C4A6. Implications for the location of a C5 binding site in C4". Journal of Immunology (Baltimore, Md.: 1950). 148 (9): 2803–2811. doi:10.4049/jimmunol.148.9.2803. PMID 1573269. S2CID 20793783.
- Kinoshita, T; Dodds, A W; Law, S K A; Inoue, K (1 August 1989). "The low C5 convertase activity of the C4A6 allotype of human complement component C4". Biochemical Journal. 261 (3): 743–748. doi:10.1042/bj2610743. PMC 1138894. PMID 2803239.
- Vogel CW, Müller-Eberhard HJ (1982). "The Cobra Venom Factor-dependentC 3 Convertase of Human Complement". J Biol Chem. 257 (14): R8292–8299. doi:10.1016/S0021-9258(18)34330-8. PMID 6919543.
- Polley MJ, Müller-Eberhard HJ (1967). "Enhancement of the Hemolytic Activity of the Second Component of Human Complement by Oxidation". J Exp Med. 126 (6): R1013–1025. doi:10.1084/jem.126.6.1013. PMC 2138419. PMID 4964564.
- Heinen S, Hartmann A, Lauer N, et al. (2009). "Factor H–related protein 1 (FHR-1) inhibits complement C5 convertase activity and terminal complex formation". Blood. 114 (12): R2439–2447. doi:10.1182/blood-2009-02-205641. PMID 19528535. S2CID 10200256.
- Ebanks, R. O.; Jaikaran, A. S.; Carroll, M. C.; Anderson, M. J.; Campbell, R. D.; Isenman, D. E. (1 May 1992). "A single arginine to tryptophan interchange at beta-chain residue 458 of human complement component C4 accounts for the defect in classical pathway C5 convertase activity of allotype C4A6. Implications for the location of a C5 binding site in C4". Journal of Immunology (Baltimore, Md.: 1950). 148 (9): 2803–2811. doi:10.4049/jimmunol.148.9.2803. PMID 1573269. S2CID 20793783.
- Kinoshita, T (1989). "The low C5 convertase activity of the C4A6 allotype of human complement component C4". Biochemical Journal. 261 (3): 743–748. doi:10.1042/bj2610743. PMC 1138894. PMID 2803239.