CREB-binding protein
CREB-binding protein, also known as CREBBP or CBP or KAT3A, (where CREB is cAMP response element-binding protein) is a coactivator encoded by the CREBBP gene in humans, located on chromosome 16p13.3.[5][6] CBP has intrinsic acetyltransferase functions; it is able to add acetyl groups to both transcription factors as well as histone lysines, the latter of which has been shown to alter chromatin structure making genes more accessible for transcription.[7][8][9][10] This relatively unique acetyltransferase activity is also seen in another transcription enzyme, EP300 (p300). Together, they are known as the p300-CBP coactivator family and are known to associate with more than 16,000 genes in humans; however, while these proteins share many structural features, emerging evidence suggests that these two co-activators may promote transcription of genes with different biological functions.[7][11][12]




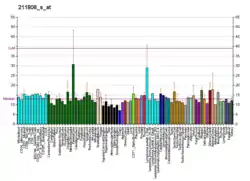
For example, CBP alone has been implicated in a wide variety of pathophysiologies including colorectal cancer as well as head and neck squamous cell carcinoma. In these diseases, association of CBP with β-catenin has been shown to promote cancer cell proliferation and disease aggressiveness, whereas p300/ β-catenin leads to cell differentiation and/ or apoptosis.[11][13] CBP has also been shown to help modulate liver function via maintenance of energy homeostasis in response to changes in cell nutrition conditions by regulating the activity of transcription factors and genes responsible for lipogenesis and gluconeogenesis.[6] CBP is also implicated in the etiologies of several other diseases including hematologic malignancies and other solid tumors, diabetes, schizophrenia, Alzheimer's disease, depression, and many other neurological conditions.[14][15][16][17][18]
Structure
The functional CBP is approximately 7362 nucleotides long and encodes 2,441 amino acids.[8][18] CBP does not directly interact with promoter elements; it is brought to the site via protein-protein interactions that are the result of its different structural domains complexing with other transcriptional co-activators.[8]
Transitional adapter zinc finger domains
CBP has two transitional adapter zinc finger (TAZ) domains that each consist of four alpha helices stabilized by zinc ions. Both TAZ1 and TAZ2 domains favor the hydrophobic residues within amphipathic amino acid sequences, and their binding affinity is enhanced by the interaction of acidic residues with positive side chains.[19] Factors that bind to TAZ2 are also potentially regulated via acetylation due to its proximity to the acetyltransferase domain (KAT).[19]

Cysteine-histidine rich regions
Although there are three cysteine-histidine rich domains identified within CBP, only domains 1 and 3 (CH1, CH3) have had their structural functions resolved.[8] A number of factors that associate with CBP bind to either the CH1 or CH3 domain, or to both, despite their locations at opposing ends of the protein. To date, little is known about the interaction between these two domains. The primary structure of CH1 and CH3 has been shown to break down into consensus sequences that are capable of chelating zinc ions (Zn2+).[8] Experiments performed also showed that the residues in these sequences are obligatory for transcriptional coactivation to occur by either CH1 or CH3.[8] The CH2 region, located in the middle of the protein, in its acetyltransferase domain, does not contain this consensus sequence, and has not been conclusively shown to bind zinc ions.
Kinase-inducible domain interacting domain
Aka KIX domain, CREB binding domain, MYB interaction domain
The kinase-inducible domain (KID) interacting domain (KIX) also is the protein domain on CBP and p300 where heterodimers form from the interaction of CBP (or p300) with other transcription factors and coactivators. It consists of three alpha and two 310 helices that have high affinity for amphipathic protein sequences.[19] Interestingly, these helices can fold into a number of different conformers which enables the domain to maintain both a level of promiscuity while also exerting regulatory control.[19] The KIX domain controls the rate of transcription and has been shown to be critical for hematopoietic differentiation.[8][19][20] Some of the proteins that bind to this domain have been shown to bind competitively—such as CREB and Myb—whereas others bind via allosteric cooperation as in the case of MLL and Myb.[19]
Bromodomain
Bromodomains (BRD) consist of approximately 110 amino acids, that function to recognize acetylated lysine molecules.[8] They consist of four left-handed alpha helices connected by loops which form hydrophobic binding pockets.[8][19] The bromodomain of CBP binds to regions of the genome are rich with acetylated lysine residues, meaning they have lost their positive charge, decreasing histone affinity for DNA, which makes the region more open and accessible for transcription. Acetylated p53 and STAT3 have been shown to bind to CBP's bromodomain.[18]
Lysine acetyltransferase domain
The 380 protein residue lysine acetyltransferase (KAT) domain is arguably one of the most important and identifying structural components of CBP. Its activity is regulated via phosphorylation of CBP. What's interesting is that it is able to acetylate not just histones but other proteins as well. Currently there are over 100 known substrates for CBP's KAT domain including proteins such as p53, E2F-1-3, GATA-1, MyoD and CREB.[18] The ability of CBP to acetylate p53 via its KAT domain, which then increases p53's affinity for CBP's bromodomain showcases an interesting mechanism by which this unique protein can self-servingly regulate gene transcription.
Nuclear receptor coactivator binding domain
Aka interferon response factor binding domain (IBiD)
The nuclear receptor coactivator binding domain (NCBD) located at the c-terminus of CBP, in its unbound state, fluctuates between multiple conformations.[19] Binding of a target protein to NCBD will cause it to fold into three helices specifically functioned to associated with the disordered domains of its binding partners.[19] Among the proteins known to bind to this region include ACTR (p160)--a coactivator for the thyroid and retinoid receptors, its homolog SRC-1, p53, and SMAD.[20][19]
Interactions
Proteins shown to interact specifically with CBP
Function
This gene is ubiquitously expressed and is involved in the transcriptional coactivation of many different transcription factors. CBP has two critical mechanisms by which it is able to regulate gene expression: as an acetyltransferase, and as a protein scaffold that helps recruit and construct the complexes that are necessary for transcription or chromatin remodeling. Phosphorylation of CBP increases its acetyltransferase activity, a process hypothesized to be regulated in a cell cycle dependent manner.[18] Recent results suggest that novel CBP-mediated post-translational N-glycosylation activity alters the conformation of CBP-interacting proteins, leading to regulation of gene expression, cell growth and differentiation,[28]
Distinction from p300
Often in scientific papers (especially in less recent ones), CBP and p300 are used interchangeably as CBP/p300. This is a reasonable amalgamation given the sequence homology, structural similarity and binding behaviors of these two proteins. However, ongoing research shows that CBP and p300 maintain distinct biological functions and should therefore not be conflated.
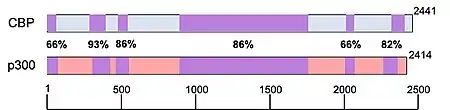
It has been reported that despite having common histone substrates, when there are lower amounts of histone or acetyl-CoA available, CBP and p300 have different, preferred acetyltransferase substrates.[18] In an experiment performed involving the Kaposi sarcoma-associated herpesvirus, the pathological protein (vIRF), was shown to be upregulated by CBP and repressed by p300.[12] p300 homozygous knockout studies in mice were embryonic lethal, with improper neurulation and poor heart development occurring during their limited survival. Additionally, the fibroblasts isolated from these mice were unable to properly proliferate and were lacking the retinoic acid receptor.[7][18] Transgenically altered mice homozygous for mutated copies of CBP (missing the KAT domain), were also embryonic lethal, however instead, these mice had poor vascular angiogenesis and abnormal hematopoiesis characterized by the absence of progenitor cell proliferation and an altered hematopoietic microenvironment.[18] The fact that both CBP and p300 homozygous knockouts were embryonic lethal suggest that these factors play a critical role in embryogenesis. The differential phenotypes of these embryos also indicate that CBP and p300 regulate different aspects of embryological development.
Role in cell cycle regulation
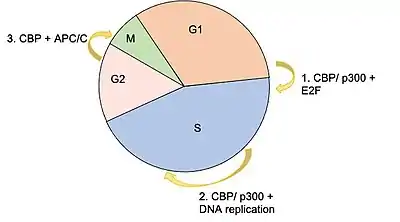
Studies performed in the late 1990s demonstrated that peak CBP acetyltransferase activity occurs at the transition between the G1/S phase of the cell cycle checkpoint.[29] Given the concentrations of CDK2 present in the cell at this stage of the cell cycle, it was hypothesized that CDK2 may be a key regulator of these post-translational modifications.[7] It turned out that administration of cyclin E/CDK2 inhibitors did in fact inhibit the enzymatic activity of CBP's KAT domain.[29] Other proteins that have been shown to phosphorylate CBP include MAP Kinase, PKA and CAMK4.[7][18] Ser-133 has been shown to be an important residue that is phosphorylated by PKA to initiate CBP transcriptional activity.[30][20]
The E2F family of transcription factors are critical for taking a cell from G1 to S phase of the cell cycle.[31] They bind to a sequence consensus in the promoter region of genes involved in DNA replication. CBP (and p300) have been shown to interact with E2F proteins as both a coactivator, and as an acetyltransferase, the latter of which causes increased E2F DNA binding affinity.[5][12] A knockout study that was published in 2000, which used a microinjection of an antibody against CBP/p300 significantly diminished the number of cells able to progress into S phase, further supporting the idea that CBP is essential in the transcription of factors necessary G1/S phase transition.[12]
CBP is also believed to facilitate the process of DNA replication during S phase by acetylating histones around origins of replication.[12] Acetylation of histones, specifically lysine residues in the histones, weakens the electric charge interaction between the histone and DNA, causing this area to become more open and accessible for the machinery required for DNA replication. Two histone acetylation markers that have been associated with active regions include histone 3 lysine 18 acetylation (H3K18ac) and histone 3 lysine 27 acetylation (H3K27ac).[12] It has also been shown that CBP acetylates two endonucleases (FEN1, DNA2) that are involved in processing Okazaki fragments.[12]
Another key component of the cell cycle that is regulated by CBP is the anaphase promoting complex/cyclosome (APC/C). This complex consists of numerous subunits that are grouped into two subdomains, the "Arc Lamp" and the "Platform," and functions as an E3 ubiquitin ligase that targets components related to the cell cycle such as cyclin B, securin, and PLK1 for proteasome degradation.[32][33] Two subunits of the APC/C have been shown to directly interact with CBP: AP5, which is located in the Platform subdomain, and AP7, located in the Arc Lamp subdomain.[32][33] Experiments performed using RNAi to completely suppress CBP and p300 showed significant increases in protein concentrations for those normally targeted by the APC/C, and caused a number of cells to become arrested in the mitotic phase of the cell cycle.[33]
CBP and p300 have been shown to acetylate key factors involved in different DNA repair processes, including base excision repair, nucleotide excision repair and non-homologous end joining.[23] CBP and p300 play a role in the acetylation of DNA damage response proteins and this post-translational modification influences their function.[23]
Role in disease
Rubinstein–Taybi syndrome
Rubinstein–Taybi syndrome (RTS) is a rare genetic disorder that is the result of genetic mutations in either CBP or p300. RTS Type 1, which is caused by CBP mutations, for which over 500 different variations have been documented, accounts for approximately 55% of all cases, whereas RTS Type 2, which is caused by any of the nearly 120 different types of p300 mutations, accounts for only 8% of diagnosed cases.[34] The majority of these mutations have been shown to cause loss of function of the gene via deletions, point or truncating mutations.[12] Statistics indicate that RTS patients have and increased risk of cancer, with approximately 5% of that attributable to pediatric malignancies originating from the neural crest.[12] Individuals with RTS frequently have skeletal abnormalities, neuroanatomical defects, and mental impairments including lower levels of intelligence, attention deficits and impaired motor coordination.[22]
| Cancer type | N | % of samples mutated |
|---|---|---|
| Follicular lymphoma | 66 | 33.3 |
| Skin squamous cell carcinoma | 77 | 28.6 |
| Marginal zone B-cell lymphoma | 15 | 13.3 |
| Diffuse large B-cell lymphoma | 242 | 12.0 |
| Salivary gland carcinoma | 63 | 9.5 |
| Bladder carcinoma | 438 | 8.9 |
| Endometrial carcinoma | 337 | 8.0 |
| Lung small-cell carcinoma | 52 | 7.7 |
| ER+ Breast carcinoma | 80 | 7.5 |
Cancer
CBP has been shown to play a role in every stage of tumor development.[8] Due to its critical role in regulation of cell proliferation, growth, migration and apoptosis, it is considered to be an oncogene, or tumor suppressor.[5] Contrarily To date, increased CBP activity has been implicated in a variety of different malignancies including breast cancer, lung cancer, prostate cancer, colorectal cancer, acute leukemias, head and neck cancer, and many others.[8][11][18][12] According to the Catalog of Somatic Mutations in Cancer (COSMIC), the most common genetic mutations in CBP are missense mutations (accounting for ~71% of all CBP mutations). The most frequent mutations occur in the KAT domain, resulting largely in either decreased or inhibited acetyltransferase activity.[12]
Hematological malignancies
Embryonic mice heterozygous for CBP (Cbp+/-) exhibited "extramedullary hematopoiesis, decreased bone marrow cellularity [a lower ratio of bone marrow to fat], and hematopoietic differentiation abnormalities."[18] By age 1, these mice had an increased incidence of leukemia or hematologic neoplasia.[25] Interestingly, tumor sequencing showed loss of heterozygosity for the wild type allele.[18] One explanation proposed for these experimental results is that CBP plays a role in hematopoietic stem cell self-renewal.[18]
In cases of patients diagnosed with Acute Myeloid Leukemia (AML) and Myelodysplastic Syndrome, CBP has been shown to gain function.[12] This occurs via chromosomal translocations between CBP and other acetyltransferase called the monocytes leukemia zinc finger (MOZ), and or between MORF (MOZ-related factor) and MLL (mixed-lineage leukemia) genes.[12] Both instances result in fused proteins in which C-terminus of CBP is lost, and the acetyltransferase domains from both proteins remain, resulting in up-regulated KAT activity and onset of disease.[12]
For patients with a relapsing case of Acute Lymphoblastic Leukemia (ALL), it was reported approximately 18% of them had CBP KAT domain mutations.[18]
Solid tumors
CBP mutations, though relatively infrequent, have been identified in lung cancer.[21] Further analysis has also revealed that during the early stages of respiratory epithelium tumorigenesis, there is increased expression in CBP as well as AP-1 and cyclin D1, factors known to be associated with CBP transcriptional activity. This over-expression may lead to downstream signaling events that favor lung tumor development.[21]
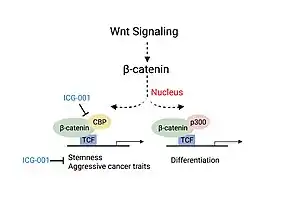
Colorectal cancer and head and neck cancer (HNSCC) disease severity have been linked to the association of CBP with β-catenin, a critical factor involved in the canonical Wnt signaling pathway.[11][13] Association of CBP with β-catenin leads to the transcription of genes responsible for more aggressive cancer traits including the presence of cancer stem cell populations, decreased immune cell infiltration and likelihood of metastasis.[13] Experiments studying the use of a small molecule inhibitor of β-catenin/CBP association (ICG-001), that does not block p300/β-catenin association, saw decreased carcinogenesis and increased cellular differentiation and apoptosis.[11][13]
Increased nuclear hormone signaling, mediated by the androgen (AR) and estrogen (ER) receptors, are responsible for a number of prostate and breast cancer cases, respectively.[11] CBP is known to interact with the AR and ER in both coactivator and acetyltransferase contexts. Inhibition of CBP KAT activity has been shown to decrease AR and ER signaling by down regulating receptor expression; this in turn suppresses tumorigenesis of both malignancies.[11]
Metabolic homeostasis
Energy homeostasis, which relies on a balance of glucose and lipids, is essential for organism survival.[6] Diseases involving impaired metabolic activity include obesity, Type 2 Diabetes (T2D) and nonalcoholic fatty liver disease (NAFLD). In the context equilibrium, over nutrition promotes lipogenesis (lipid synthesis) in response to increased glucose and insulin concentrations, and fasting promotes β-oxidation (lipid breakdown) and gluconeogenesis (synthesis of glucose).[6] Experiments performed in diet-induced obese mice saw increased lipogenesis driven by over expression of sterol regulatory element binding protein 1C (SREBP1C), which works in coordination with the carbohydrate responsive-element binding protein (ChREBP). Both are transcription factors critical for lipogenesis, and both are acetylated by CBP, a post translational modification that increases their transcriptional activity.[6] To balance the increase in lipid synthesis, the body needs to be able to export the macromolecule out of cells and storage. Microsomal triglyceride transfer protein (MTP) is responsible for lipoprotein assembly and secretion, and it associates with an RNA helicase, DDX3, which interacts with CBP causing HNF4 acetylation, which in turn increases the rate of transcription of MTP in a positive feedback loop.[6]
CBP also has a role in regulation of glucose homeostasis during fasting conditions. Research has also shown that glucagon, a hormone released when the body has low blood sugar, activates cAMP response element binding protein (CREB), which then binds to CBP as a coactivator for transcription of FOXO1.[6] FOXO1 is a transcription factor for enzymes including glucose-6-phosphatase and phosphoenolpyruvate carboxykinase (PECK1), which are necessary for gluconeogenesis.
Neurological disorders
CREB has been shown to have neuroprotective properties.[14] Because of its association with CBP, understanding the role of CBP in neurological pathways and how aberrations influence disease is becoming of increasing interest. Numerous animal models have been designed in order to evaluate changes in motor, learning and memory function in mice with CBP mutations. Conditional knockout (cKO) mice that were hemizygous for CBP or had CBP point mutations exhibited memory defects—specifically related to long-term memory.[22] For mice with homozygous point mutations in their CBP KIX domain, they demonstrated impaired motor skill learning and execution.[22] In conjunction with the neurological challenges experienced by RTS patients (lower levels of intelligence, attention deficits), CBP is attributable to a variety of neurological symptoms characteristic of many different types of diseases.
Fetal alcohol spectrum disorders
Fetal alcohol spectrum disorders (FASD) is a classification of diseases that all result from alcohol exposure during pregnancy.[22] Symptoms of these disorders include poor cerebellar-dependent learning, motor coordination and impaired balance.[22] In rats with FASD, it was shown that they had decreased concentrations of CBP and lower amounts of H3 and H4 acetylation.[22]
Huntington's disease
Huntington's disease (HD) is a fatal, progressing neurodegenerative disorder that is the result of a genetic mutation in the Huntingtin gene causing synthesis of a mutated huntingtin (Htt) protein.[22] Symptoms most frequently associated with this disease are movement disorders, including impaired motor function, behavioral modification and cognitive impairment that ultimately results in dementia.[35] It has been observed in animal models that HD subjects had diminished CBP activity and decreased neuronal histone acetylation.[22] Research has shown that mutHtt directly interacts with CBP It has been hypothesized that mutant Htt is either capable of degrading CBP, or it directly inhibits CBP's acetyltransferase domain.[22][14]
Alzheimer's disease
Alzheimer's disease (AD) is a progressive neurodegenerative disease whose pathology is diagnosed based on the presence of neuritic amyloid beta (Aβ) plaques and neurofibrillary tau (τ) tangles.[22] Because the exact causes of the disease are not clearly understood, there are a number of different mechanisms by which CBP is hypothesized to play a role in the progression of AD. In many cases of early-onset familial AD (FAD), there are mutations of the proteins that make up the enzyme responsible for the creation of Aβ plaques. CBP activity is decreased in the absence of these proteins (presenilin 1 or presenilin 2).[16][22] Additionally, in mouse models of AD, it has been shown that there is a decrease in neuronal histone acetylation, a critical function of CBP.[22]
Inhibition of CBP
Given the control CBP has over a wide variety of physiological processes, the development of inhibitors for CBP activity have become increasingly important as potential therapies. To date, only a fraction of what has been discovered have progressed into clinical trials.
| Drug | Stage of development | Disease(s)/ areas targeted | Reference |
|---|---|---|---|
| A-485 | In vitro/ in vivo | Hematological malignancies, androgen receptor positive
prostate cancer |
[36] |
| C646 | In vitro/ in vivo | Solid tumors, neuroepithelial histone acetylation rescue | [8] |
| CBP30 | In vitro | Autoimmune diseases | [8] |
| CCS1477 | Phase 1b/2a | Advanced castration resistant prostate cancer,
hematological malignancies |
[10][8] |
| CPI-637 | In vitro/ in vivo | Castration resistant prostate cancer | [37] |
| dCBP-1 | In silicon | Multiple myeloma (MM) | [38] |
| DC_CP20 | In silicon | Leukemia | [8] |
| E7386 | Phase 1 | Solid tumors, prevents β-catenin/CBP protein-protein
interactions |
[39] |
| Garcinol | In vitro/ in vivo | Esophageal cancer | [36] |
| GNE-049 | In vitro/ in vivio | Breast and prostate cancer | [38] |
| GNE-207 | In vitro/ in vivo | Acute myeloid leukemia (AML), potential other
hematological malignancies |
[8] |
| GNE-781 | In vitro/ in vivo | Acute myeloid leukemia (AML), potential other
hematological malignancies, prostate and breast cancer |
[38][8][40] |
| HBS1 | In vitro/ in vivo | Renal cell carcinoma | [8][41] |
| I-CBP112 | In vitro/ in vivo | CBP bromodomain | [8] |
| ICG-001 | In vitro/ in vivo | Colorectal cancer, head and neck squamous cell carcinoma | [11][13] |
| KCN1 | In vitro/ in vivo | Glioma | [8][42] |
| MYBMIM | In silicon | Acute myeloid leukemia (AML), prevents CBP/Myb
protein-protein interactions |
[8] |
| NASTRp | In vitro/ in vivo | Lung adenocarcinoma | [8] |
| NEO2734 | In vitro/ in vivo | Prostate cancer | [8] |
| Nicur | In silicon | Gastrointestinal cacner | [8] |
| OHM1 | In vitro/ in vivo | Prevents CBP/HIF-1α protein-protein interaction | [8] |
| PRI-724 | In vitro/ in vivo | Liver fibrosis | [6] |
| PU139 | In vitro/ in vivo | Neuroblastoma | [36] |
| Y08197 | In vitro | Castration resistant prostate cancer | [37] |
References
- GRCh38: Ensembl release 89: ENSG00000005339 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000022521 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Chan HM, La Thangue NB (July 2001). "p300/CBP proteins: HATs for transcriptional bridges and scaffolds". Journal of Cell Science. 114 (Pt 13): 2363–2373. doi:10.1242/jcs.114.13.2363. PMID 11559745.
- Yao W, Wang T, Huang F (2018). "p300/CBP as a Key Nutritional Sensor for Hepatic Energy Homeostasis and Liver Fibrosis". BioMed Research International. 2018: 8168791. doi:10.1155/2018/8168791. PMC 5976926. PMID 29862292.
- Vo N, Goodman RH (April 2001). "CREB-binding protein and p300 in transcriptional regulation". The Journal of Biological Chemistry. 276 (17): 13505–13508. doi:10.1074/jbc.R000025200. PMID 11279224.
- Akinsiku OE, Soremekun OS, Soliman ME (February 2021). "Update and Potential Opportunities in CBP [Cyclic Adenosine Monophosphate (cAMP) Response Element-Binding Protein (CREB)-Binding Protein] Research Using Computational Techniques". The Protein Journal. 40 (1): 19–27. doi:10.1007/s10930-020-09951-8. PMC 7868315. PMID 33394237.
- Guo P, Chen W, Li H, Li M, Li L (October 2018). "The Histone Acetylation Modifications of Breast Cancer and their Therapeutic Implications". Pathology & Oncology Research. 24 (4): 807–813. doi:10.1007/s12253-018-0433-5. PMID 29948617. S2CID 47020435.
- He ZX, Wei BF, Zhang X, Gong YP, Ma LY, Zhao W (January 2021). "Current development of CBP/p300 inhibitors in the last decade". European Journal of Medicinal Chemistry. 209: 112861. doi:10.1016/j.ejmech.2020.112861. ISSN 0223-5234. PMID 33045661. S2CID 222319204.
- Bordonaro M, Lazarova DL (July 2015). "CREB-binding protein, p300, butyrate, and Wnt signaling in colorectal cancer". World Journal of Gastroenterology. 21 (27): 8238–8248. doi:10.3748/wjg.v21.i27.8238. PMC 4507093. PMID 26217075.
- Attar N, Kurdistani SK (March 2017). "Exploitation of EP300 and CREBBP Lysine Acetyltransferases by Cancer". Cold Spring Harbor Perspectives in Medicine. 7 (3): a026534. doi:10.1101/cshperspect.a026534. PMC 5334244. PMID 27881443.
- Kartha VK, Alamoud KA, Sadykov K, Nguyen BC, Laroche F, Feng H, et al. (July 2018). "Functional and genomic analyses reveal therapeutic potential of targeting β-catenin/CBP activity in head and neck cancer". Genome Medicine. 10 (1): 54. doi:10.1186/s13073-018-0569-7. PMC 6053793. PMID 30029671.
- Rouaux C, Loeffler JP, Boutillier AL (September 2004). "Targeting CREB-binding protein (CBP) loss of function as a therapeutic strategy in neurological disorders". Biochemical Pharmacology. 68 (6): 1157–1164. doi:10.1016/j.bcp.2004.05.035. PMID 15313413.
- Wang H, Xu J, Lazarovici P, Quirion R, Zheng W (2018-08-30). "cAMP Response Element-Binding Protein (CREB): A Possible Signaling Molecule Link in the Pathophysiology of Schizophrenia". Frontiers in Molecular Neuroscience. 11: 255. doi:10.3389/fnmol.2018.00255. PMC 6125665. PMID 30214393.
- Amidfar M, de Oliveira J, Kucharska E, Budni J, Kim YK (September 2020). "The role of CREB and BDNF in neurobiology and treatment of Alzheimer's disease". Life Sciences. 257: 118020. doi:10.1016/j.lfs.2020.118020. PMID 32603820. S2CID 220287306.
- Steven A, Friedrich M, Jank P, Heimer N, Budczies J, Denkert C, Seliger B (October 2020). "What turns CREB on? And off? And why does it matter?". Cellular and Molecular Life Sciences. 77 (20): 4049–4067. doi:10.1007/s00018-020-03525-8. PMC 7532970. PMID 32347317.
- Dutta R, Tiu B, Sakamoto KM (September 2016). "CBP/p300 acetyltransferase activity in hematologic malignancies". Molecular Genetics and Metabolism. 119 (1–2): 37–43. doi:10.1016/j.ymgme.2016.06.013. PMID 27380996.
- Dyson HJ, Wright PE (March 2016). "Role of Intrinsic Protein Disorder in the Function and Interactions of the Transcriptional Coactivators CREB-binding Protein (CBP) and p300". The Journal of Biological Chemistry. 291 (13): 6714–6722. doi:10.1074/jbc.R115.692020. PMC 4807259. PMID 26851278.
- Sapio L, Salzillo A, Ragone A, Illiano M, Spina A, Naviglio S (October 2020). "Targeting CREB in Cancer Therapy: A Key Candidate or One of Many? An Update". Cancers. 12 (11): 3166. doi:10.3390/cancers12113166. PMC 7693618. PMID 33126560.
- Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG (April 2007). "Roles of CREB-binding protein (CBP)/p300 in respiratory epithelium tumorigenesis". Cell Research. 17 (4): 324–332. doi:10.1038/cr.2007.10. PMID 17372613. S2CID 36084602.
- Valor LM, Viosca J, Lopez-Atalaya JP, Barco A (2013-07-31). "Lysine acetyltransferases CBP and p300 as therapeutic targets in cognitive and neurodegenerative disorders". Current Pharmaceutical Design. 19 (28): 5051–5064. doi:10.2174/13816128113199990382. PMC 3722569. PMID 23448461.
- Dutto I, Scalera C, Prosperi E (April 2018). "CREBBP and p300 lysine acetyl transferases in the DNA damage response". Cellular and Molecular Life Sciences. 75 (8): 1325–1338. doi:10.1007/s00018-017-2717-4. PMID 29170789. S2CID 3951961.
- Mullan PB, Quinn JE, Harkin DP (September 2006). "The role of BRCA1 in transcriptional regulation and cell cycle control". Oncogene. 25 (43): 5854–5863. doi:10.1038/sj.onc.1209872. PMID 16998500. S2CID 41157893.
- Blobel GA (2000-02-01). "CREB-binding protein and p300: molecular integrators of hematopoietic transcription". Blood. 95 (3): 745–755. doi:10.1182/blood.V95.3.745.003k05_745_755. ISSN 1528-0020. PMID 10648382.
- Newton AL, Sharpe BK, Kwan A, Mackay JP, Crossley M (May 2000). "The transactivation domain within cysteine/histidine-rich region 1 of CBP comprises two novel zinc-binding modules". The Journal of Biological Chemistry. 275 (20): 15128–15134. doi:10.1074/jbc.M910396199. PMID 10748221.
- Cantor AB, Orkin SH (May 2002). "Transcriptional regulation of erythropoiesis: an affair involving multiple partners". Oncogene. 21 (21): 3368–3376. doi:10.1038/sj.onc.1205326. PMID 12032775. S2CID 33364155.
- Siddique H, Rao VN, Reddy ES (August 2009). "CBP-mediated post-translational N-glycosylation of BRCA2". International Journal of Oncology. 35 (2): 387–391. doi:10.3892/ijo_00000351. PMID 19578754.
- Ait-Si-Ali S, Ramirez S, Barre FX, Dkhissi F, Magnaghi-Jaulin L, Girault JA, et al. (November 1998). "Histone acetyltransferase activity of CBP is controlled by cycle-dependent kinases and oncoprotein E1A". Nature. 396 (6707): 184–186. Bibcode:1998Natur.396..184A. doi:10.1038/24190. PMID 9823900. S2CID 4340781.
- Zhang H, Kong Q, Wang J, Jiang Y, Hua H (November 2020). "Complex roles of cAMP-PKA-CREB signaling in cancer". Experimental Hematology & Oncology. 9 (1): 32. doi:10.1186/s40164-020-00191-1. PMC 7684908. PMID 33292604.
- Rubin SM, Sage J, Skotheim JM (October 2020). "Integrating Old and New Paradigms of G1/S Control". Molecular Cell. 80 (2): 183–192. doi:10.1016/j.molcel.2020.08.020. PMC 7582788. PMID 32946743.
- Bodrug T, Welsh KA, Hinkle M, Emanuele MJ, Brown NG (2021-05-24). "Intricate Regulatory Mechanisms of the Anaphase-Promoting Complex/Cyclosome and Its Role in Chromatin Regulation". Frontiers in Cell and Developmental Biology. 9: 687515. doi:10.3389/fcell.2021.687515. PMC 8182066. PMID 34109183.
- Turnell AS, Stewart GS, Grand RJ, Rookes SM, Martin A, Yamano H, et al. (December 2005). "The APC/C and CBP/p300 cooperate to regulate transcription and cell-cycle progression". Nature. 438 (7068): 690–695. Bibcode:2005Natur.438..690T. doi:10.1038/nature04151. PMID 16319895. S2CID 4432156.
- Van Gils J, Magdinier F, Fergelot P, Lacombe D (June 2021). "Rubinstein-Taybi Syndrome: A Model of Epigenetic Disorder". Genes. 12 (7): 968. doi:10.3390/genes12070968. PMC 8303114. PMID 34202860.
- Kim C, Yousefian-Jazi A, Choi SH, Chang I, Lee J, Ryu H (November 2021). "Non-Cell Autonomous and Epigenetic Mechanisms of Huntington's Disease". International Journal of Molecular Sciences. 22 (22): 12499. doi:10.3390/ijms222212499. PMC 8617801. PMID 34830381.
- Wang Y, Xie Q, Tan H, Liao M, Zhu S, Zheng LL, et al. (November 2021). "Targeting cancer epigenetic pathways with small-molecule compounds: Therapeutic efficacy and combination therapies". Pharmacological Research. 173: 105702. doi:10.1016/j.phrs.2021.105702. PMID 34102228. S2CID 235378858.
- Akinsiku OE, Soremekun OS, Olotu FA, Soliman ME (2020). "Exploring the Role of Asp1116 in Selective Drug Targeting of CREBcAMP- Responsive Element-binding Protein Implicated in Prostate Cancer". Combinatorial Chemistry & High Throughput Screening. 23 (3): 178–184. doi:10.2174/1386207323666200219122057. PMID 32072894. S2CID 211193693.
- Waddell AR, Huang H, Liao D (June 2021). "CBP/p300: Critical Co-Activators for Nuclear Steroid Hormone Receptors and Emerging Therapeutic Targets in Prostate and Breast Cancers". Cancers. 13 (12): 2872. doi:10.3390/cancers13122872. PMC 8229436. PMID 34201346.
- "The CREB-binding protein (CBP)/β-catenin inhibitor E7386, co-created by Eisai and PRISM BioLab, achieved the clinical POC (Proof of Concept) | News Release:2021 | Eisai Co., Ltd". www.eisai.com. Retrieved 2021-12-04.
- Romero FA, Murray J, Lai KW, Tsui V, Albrecht BK, An L, et al. (November 2017). "GNE-781, A Highly Advanced Potent and Selective Bromodomain Inhibitor of Cyclic Adenosine Monophosphate Response Element Binding Protein, Binding Protein (CBP)". Journal of Medicinal Chemistry. 60 (22): 9162–9183. doi:10.1021/acs.jmedchem.7b00796. PMID 28892380.
- Farria A, Li W, Dent SY (September 2015). "KATs in cancer: functions and therapies". Oncogene. 34 (38): 4901–4913. doi:10.1038/onc.2014.453. PMC 4530097. PMID 25659580.
- Yin S, Kaluz S, Devi NS, Jabbar AA, de Noronha RG, Mun J, et al. (December 2012). "Arylsulfonamide KCN1 inhibits in vivo glioma growth and interferes with HIF signaling by disrupting HIF-1α interaction with cofactors p300/CBP". Clinical Cancer Research. 18 (24): 6623–6633. doi:10.1158/1078-0432.CCR-12-0861. PMC 3518680. PMID 22923450.
Further reading
- Goldman PS, Tran VK, Goodman RH (1997). "The multifunctional role of the co-activator CBP in transcriptional regulation". Recent Progress in Hormone Research. 52: 103–19, discussion 119–20. PMID 9238849.
- Marcello A, Zoppé M, Giacca M (March 2001). "Multiple modes of transcriptional regulation by the HIV-1 Tat transactivator". IUBMB Life. 51 (3): 175–181. doi:10.1080/152165401753544241. PMID 11547919. S2CID 10931640.
- Matt T (2002). "Transcriptional control of the inflammatory response: a role for the CREB-binding protein (CBP)". Acta Medica Austriaca. 29 (3): 77–79. doi:10.1046/j.1563-2571.2002.02010.x. PMID 12168567.
- Combes R, Balls M, Bansil L, Barratt M, Bell D, Botham P, et al. (2002). "An assessment of progress in the use of alternatives in toxicity testing since the publication of the report of the second FRAME Toxicity Committee (1991)". Alternatives to Laboratory Animals. 30 (4): 365–406. doi:10.1177/026119290203000403. PMID 12234245. S2CID 26326825.
- Minghetti L, Visentin S, Patrizio M, Franchini L, Ajmone-Cat MA, Levi G (May 2004). "Multiple actions of the human immunodeficiency virus type-1 Tat protein on microglial cell functions". Neurochemical Research. 29 (5): 965–978. doi:10.1023/B:NERE.0000021241.90133.89. PMID 15139295. S2CID 25323034.
- Kino T, Pavlakis GN (April 2004). "Partner molecules of accessory protein Vpr of the human immunodeficiency virus type 1". DNA and Cell Biology. 23 (4): 193–205. doi:10.1089/104454904773819789. PMID 15142377.
- Greene WC, Chen LF (2004). "Regulation of NF-kappaB action by reversible acetylation". Novartis Foundation Symposium. Novartis Foundation Symposia. 259: 208–17, discussion 218–25. doi:10.1002/0470862637.ch15. ISBN 9780470862612. PMID 15171256.
- Liou LY, Herrmann CH, Rice AP (September 2004). "HIV-1 infection and regulation of Tat function in macrophages". The International Journal of Biochemistry & Cell Biology. 36 (9): 1767–1775. doi:10.1016/j.biocel.2004.02.018. PMID 15183343.
- Pugliese A, Vidotto V, Beltramo T, Petrini S, Torre D (2005). "A review of HIV-1 Tat protein biological effects". Cell Biochemistry and Function. 23 (4): 223–227. doi:10.1002/cbf.1147. PMID 15473004. S2CID 8408278.
- Bannwarth S, Gatignol A (January 2005). "HIV-1 TAR RNA: the target of molecular interactions between the virus and its host". Current HIV Research. 3 (1): 61–71. doi:10.2174/1570162052772924. PMID 15638724.
- Le Rouzic E, Benichou S (February 2005). "The Vpr protein from HIV-1: distinct roles along the viral life cycle". Retrovirology. 2: 11. doi:10.1186/1742-4690-2-11. PMC 554975. PMID 15725353.
- Gibellini D, Vitone F, Schiavone P, Re MC (April 2005). "HIV-1 tat protein and cell proliferation and survival: a brief review". The New Microbiologica. 28 (2): 95–109. PMID 16035254.
- Hetzer C, Dormeyer W, Schnölzer M, Ott M (October 2005). "Decoding Tat: the biology of HIV Tat posttranslational modifications". Microbes and Infection. 7 (13): 1364–1369. doi:10.1016/j.micinf.2005.06.003. PMID 16046164.
- Peruzzi F (January 2006). "The multiple functions of HIV-1 Tat: proliferation versus apoptosis". Frontiers in Bioscience. 11: 708–717. doi:10.2741/1829. PMID 16146763. S2CID 12438136.
External links
- GeneReviews/NCBI/NIH/UW entry on Rubinstein-Taybi Syndrome
- CREBBP+protein,+human at the U.S. National Library of Medicine Medical Subject Headings (MeSH)
- NURSA C39
- Drosophila nejire - The Interactive Fly
- Human CREBBP genome location and CREBBP gene details page in the UCSC Genome Browser.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.
PDB gallery | |
|---|---|
|