CITE-Seq
CITE-Seq (Cellular Indexing of Transcriptomes and Epitopes by Sequencing) is a method for performing RNA sequencing along with gaining quantitative and qualitative information on surface proteins with available antibodies on a single cell level.[1] So far, the method has been demonstrated to work with only a few proteins per cell. As such, it provides an additional layer of information for the same cell by combining both proteomics and transcriptomics data. For phenotyping, this method has been shown to be as accurate as flow cytometry (a gold standard) by the groups that developed it.[2] It is currently one of the main methods, along with REAP-Seq, to evaluate both gene expression and protein levels simultaneously in different species.
The method was established by the New York Genome Center in collaboration with the Satija lab.[2], while a similar approach was earlier shown by AbVitro Inc..
Applications
Concurrent measurement of both protein and transcript levels opens up opportunities to use CITE-Seq in various biological areas, some of which were touched upon by the developers. For instance, it may be used to characterize tumor heterogeneity in different cancers, a major research field.[3] It also permits identifying rare subpopulations of cells as a high-throughput single-cell method and thus detect information otherwise lost with bulk methods.[3] It also may aid in tumor classification - for example, identification of novel subtypes.[3] All of the above are possible due to single-cell output of both protein and transcript data at the same time, also leading to novel information on protein-RNA correlation.
It also has potential in immunology. For example, it can be utilized for immune cell characterization – recent research on T-cells has investigated the ability of T cells to maintain an effector state.[4] Another study by one of CITE-Seq coauthors suggested CITE-Seq as a methods to look at the mechanisms of host-pathogen interactions.[5]
Workflow
CITE-seq, like any other sequencing technique, has a wet lab portion, where the actual antibodies are prepared, cells stained, cDNA synthesized and RNA libraries are prepared that are further sequenced, and a dry lab portion for analysis of the sequencing data obtained. The most crucial part in the wet lab experiments is designing the antibody-oligonucleotide conjugates and titrating the amount of each conjugate that needs to be present in the pool to achieve a desired read-out and quantification.

Wet lab workflow
The first step involves preparation of the antibody-oligo conjugates also known as Antibody-Derived Tags (ADTs). ADT preparation involves labeling an antibody directed against a cell surface protein of interest with oligonucleotides for barcoding the antibody.
Once you have the ADTs, the next step is to bind the cells with the desired ADT pool. The scRNA-seq libraries can be prepared using Drop-seq, 10X Genomics or ddSeq methods. In brief, ADT labelled cells are encapsulated within a droplet as single cells with DNA-barcoded microbeads.[6]
Within a droplet, the cells are next lysed to release both bound ADTs as well as mRNA. These then are converted to cDNA. Each DNA sequence on a microbead has a unique barcode thus indexing cDNA with cell barcodes. cDNA is prepared from both ADTs and cellular mRNAs.
In the next step, based on the developer's guidelines, cDNA is PCR-amplified and ADT cDNA and mRNA cDNA are separated based on size (generally, ADT-derived cDNAs are < 180bp and mRNA-derived cDNAs are > 300bp).[7] Each of the separated cDNA molecules is independently amplified and purified to prepare sequencing libraries. Finally, the independent libraries are pooled together and sequenced. Thus, proteomics and transcriptomics data can be obtained from a single sequencing run.

Dry lab workflow
Analysis of single-cell sequencing presents many challenges, such as determining the best way to normalize the data.[8] Due to a new level of complications that arise from sequencing of both proteins and transcripts at a single-cell level, the developers of CITE-Seq and their collaborators are maintaining several tools to help with data analysis.
scRNA-Seq data analysis based on the developer's guidelines:[2][9] The initial analysis steps are the same as in a standard scRNA-Seq experiment. Firstly, reads need to be aligned to a reference genome of a species of interest and cells with very low number of transcripts mapped to the reference are removed. Finally, a normalized count matrix with gene expression values is obtained.
ADT data analysis[2][7][10][11] (based on the developer's guidelines): CITE-seq-Count is a Python package from CITE-Seq developers that can be used to obtain raw counts. Seurat package from Satija lab further allows combining of the protein and RNA counts and performing clustering on both measurements, as well as doing differential expression analysis between cell clusters of interest. ADT quantification needs to take into account the differences between the antibodies. Additionally, filtering may be required to reduce noise, similarly to scRNA-Seq analysis. But in contrast to RNA data, due to higher amounts of protein in a cell, there is less dropout.
The analyses may result in identification of novel cell clusters through such methods as PCA or tSNE, crucial genes responsible for a specific cell function and other new knowledge specific to a question of interest. In general, the results obtained with ADT counts substantially increase the amount of information obtained through single cell transcriptomics.
Adaptations of the technique
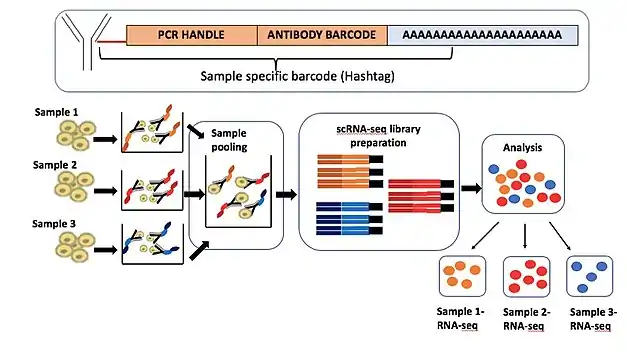
The applications of antibody-oligonucleotide conjugates have expanded beyond CITE-seq, and can be adapted for sample multiplexing as well as CRISPR screens.
Cell Hashing: New York Genome Center further adapted the use of their antibody-oligonucleotide conjugates to enable sample multiplexing for scRNA-seq. This technique called, Cell Hashing,[12] uses oligonucleotide-labelled antibodies against ubiquitously expressed cell surface proteins from a particular tissue sample. In this case, an oligonucleotide sequence contains a unique barcode which would be specific to cells from distinct samples. This sample-specific cell tagging allows pooling of the sequencing libraries prepared from different samples on a sequencing platform. Sequencing the antibody tags along with the cellular transcriptome helps identify a sample of origin for each analyzed cell. A unique barcode sequence used on the cell hashing antibody can be designed to be different from an antibody barcode present on the ADTs used in CITE-seq. This makes it possible to couple cell hashing with CITE-seq on a single sequencing run.[12] Cell hashing allows super-loading of the scRNA-seq platform, resulting in a lower cost of sequencing. It also enables detection of artifactual signals from multiplets, a major challenge in scRNA-seq. The cell hashing method has further been used by Gaublomme et al. to multiplex single-nucleus RNA-seq (snRNA-seq) by performing nucleus hashing.[13]
ECCITE-seq: Expanded CRISPR-compatible Cellular Indexing of Transcriptomes and Epitopes by sequencing or ECCITE-seq was developed to apply the use of CITE-seq to characterize multiple modalities from a single cell. By modifying the basic CITE-seq protocol to a 5’ tag-based scRNA-seq assay, it can detect transcriptome, immune receptor clonotypes, surface markers, sample identity and single guide RNAs (sgRNAs) from each single cell.[14] The ability of ECCITE-seq to detect sgRNA molecules and measure their effect on gene expression levels opens a prospect of applying this technique in CRISPR screens.
Advantages and Limitations of CITE-seq
Advantages: CITE-seq enables simultaneous analysis of the transcriptome as well as the proteome of single cells. Previous efforts of coupling index-sorting measurements from single cell sorts with scRNA-seq were limited to running a small sample size and were not compatible with multiplexing and massive parallel high-throughput sequencing. CITE-seq has been shown to be compatible with high-throughput microfluidic platforms like 10X Genomics and Drop-seq. It is also adaptable to micro/nano-well platforms. Coupling it with cell hashing enables the application of CITE-seq on bulk samples and sample multiplexing. These techniques work to reduce an overall cost of high-throughput sequencing on multiple samples. Lastly, CITE-seq can be adapted to detect small molecules, RNA interference, CRISPR, and other gene editing techniques.
Limitations: One of the limitations of CITE-Seq is a loss of location information. Due to the way the cells are treated, the spatial distribution of cells within a sample, as well as proteins within a cell is not known.[15][9] In addition, this method shares the challenges of scRNA-Seq, such as high amount of noise and possible challenges in detecting lowly expressed genes.[9] In terms of phenotyping, optimization of the assay and antibodies also presents a potential problem if proteins of interest are not included in the currently available panels.[16] Moreover, right now CITE-Seq is not able to detect intracellular proteins.[16] With the current protocol, there are many challenges that would arise during the permeabilization step, thus limiting the technique to surface markers.
Alternative methods
- REAP-seq: Peterson et al. from Merck developed a technique similar to CITE-seq called RNA Expression and Protein Sequencing assay (REAP-seq). While REAP-seq, similarly to CITE-seq, measures levels of both transcripts and proteins in a single cell, the difference between the two techniques is how the antibody is conjugated to the oligonucleotides. CITE-seq typically links the oligonucleotide to the antibody non-covalently, via streptavidin conjugation to the antibody and biotin conjugation to the oligonucleotide. REAP-seq covalently links the antibody and an aminated DNA barcode[17]
- PLAYR: PLAYR or Proximal Ligation Assay for RNA makes use of mass spectrometry to simultaneously analyse the transcriptome and protein levels in single cells. In this technique both the proteins and RNA transcripts are labelled with isotope-conjugated antibodies and isotope-labelled probes, respectively, enabling their detection on a mass spectrometer[18]
References
- Mercatelli, Daniele; Balboni, Nicola; De Giorgio, Francesca; Aleo, Emanuela; Garone, Caterina; Giorgi, Fedrico M. (2021-05-06). "The Transcriptome of SH-SY5Y at Single-Cell Resolution: A CITE-Seq Data Analysis Workflow". Methods and Protocols. 4 (2): 28. doi:10.3390/mps4020028. ISSN 2409-9279. PMC 8163004. PMID 34066513.
- Stoeckius, Marlon; Hafemeister, Christoph; Stephenson, William; Houck-Loomis, Brian; Chattopadhyay, Pratip K; Swerdlow, Harold; Satija, Rahul; Smibert, Peter (2017-07-31). "Simultaneous epitope and transcriptome measurement in single cells". Nature Methods. 14 (9): 865–868. doi:10.1038/nmeth.4380. ISSN 1548-7091. PMC 5669064. PMID 28759029.
- Tirosh, Itay; Suvà, Mario L. (2018-11-16). "Deciphering Human Tumor Biology by Single-Cell Expression Profiling". Annual Review of Cancer Biology. 3 (1): 151–166. doi:10.1146/annurev-cancerbio-030518-055609. ISSN 2472-3428. S2CID 53969464.
- Gutierrez-Arcelus, Maria; Teslovich, Nikola; Mola, Alex R.; Polidoro, Rafael B.; Nathan, Aparna; Kim, Hyun; Hannes, Susan; Slowikowski, Kamil; Watts, Gerald F. M. (2019-02-08). "Lymphocyte innateness defined by transcriptional states reflects a balance between proliferation and effector functions". Nature Communications. 10 (1): 687. Bibcode:2019NatCo..10..687G. doi:10.1038/s41467-019-08604-4. ISSN 2041-1723. PMC 6368609. PMID 30737409.
- Chattopadhyay, Pratip K.; Roederer, Mario; Bolton, Diane L. (2018-11-06). "A deadly dance: the choreography of host–pathogen interactions, as revealed by single-cell technologies". Nature Communications. 9 (1): 4638. Bibcode:2018NatCo...9.4638C. doi:10.1038/s41467-018-06214-0. ISSN 2041-1723. PMC 6219517. PMID 30401874.
- Macosko, Evan Z.; Basu, Anindita; Satija, Rahul; Nemesh, James; Shekhar, Karthik; Goldman, Melissa; Tirosh, Itay; Bialas, Allison R.; Kamitaki, Nolan (May 2015). "Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets". Cell. 161 (5): 1202–1214. doi:10.1016/j.cell.2015.05.002. ISSN 0092-8674. PMC 4481139. PMID 26000488.
- "CITE-seq". CITE-seq. Retrieved 2019-02-27.
- Gao, Shan (2018), "Data Analysis in Single-Cell Transcriptome Sequencing", Computational Systems Biology, Methods in Molecular Biology, vol. 1754, Springer New York, pp. 311–326, doi:10.1007/978-1-4939-7717-8_18, ISBN 9781493977161, PMID 29536451
- Liu, Serena; Trapnell, Cole (2016-02-17). "Single-cell transcriptome sequencing: recent advances and remaining challenges". F1000Research. 5: 182. doi:10.12688/f1000research.7223.1. ISSN 2046-1402. PMC 4758375. PMID 26949524.
- Roelli, Patrick (2019-02-23), Small script that allows to count TAGS from a CITE-seq experiment: Hoohm/CITE-seq-Count, retrieved 2019-02-27
- "Seurat". satijalab.org. Retrieved 2019-02-27.
- Stoeckius, Marlon; Zheng, Shiwei; Houck-Loomis, Brian; Hao, Stephanie; Yeung, Bertrand; Smibert, Peter; Satija, Rahul (2017-12-21). "Cell "hashing" with barcoded antibodies enables multiplexing and doublet detection for single cell genomics". Genome Biology. 19 (1): 224. doi:10.1101/237693. PMC 6300015. PMID 30567574.
- Gaublomme, Jellert T.; Li, Bo; McCabe, Cristin; Knecht, Abigail; Drokhlyansky, Eugene; Van Wittenberghe, Nicholas; Waldman, Julia; Dionne, Danielle; Nguyen, Lan (2018-11-23). "Nuclei multiplexing with barcoded antibodies for single-nucleus genomics". bioRxiv. doi:10.1101/476036. hdl:1721.1/125028.
- Mimitou, Eleni; Cheng, Anthony; Montalbano, Antonino; Hao, Stephanie; Stoeckius, Marlon; Legut, Mateusz; Roush, Timothy; Herrera, Alberto; Papalexi, Efthymia (2018-11-08). "Expanding the CITE-seq tool-kit: Detection of proteins, transcriptomes, clonotypes and CRISPR perturbations with multiplexing, in a single assay". bioRxiv. doi:10.1101/466466.
- An, Xingyue; Varadarajan, Navin (March 2018). "Single-cell technologies for profiling T cells to enable monitoring of immunotherapies". Current Opinion in Chemical Engineering. 19: 142–152. doi:10.1016/j.coche.2018.01.003. ISSN 2211-3398. PMC 6530921. PMID 31131208.
- Baron, Maayan; Yanai, Itai (2017-08-24). "New skin for the old RNA-Seq ceremony: the age of single-cell multi-omics". Genome Biology. 18 (1): 159. doi:10.1186/s13059-017-1300-5. ISSN 1474-760X. PMC 5571565. PMID 28837001.
- Peterson, Vanessa M; Zhang, Kelvin Xi; Kumar, Namit; Wong, Jerelyn; Li, Lixia; Wilson, Douglas C; Moore, Renee; McClanahan, Terrill K; Sadekova, Svetlana (2017-08-30). "Multiplexed quantification of proteins and transcripts in single cells". Nature Biotechnology. 35 (10): 936–939. doi:10.1038/nbt.3973. ISSN 1087-0156. PMID 28854175. S2CID 205285357.
- Frei, Andreas P; Bava, Felice-Alessio; Zunder, Eli R; Hsieh, Elena W Y; Chen, Shih-Yu; Nolan, Garry P; Gherardini, Pier Federico (2016-01-25). "Highly multiplexed simultaneous detection of RNAs and proteins in single cells". Nature Methods. 13 (3): 269–275. doi:10.1038/nmeth.3742. ISSN 1548-7091. PMC 4767631. PMID 26808670.