CS23D
CS23D is a web server to generate 3D structural models from NMR chemical shifts.[1] CS23D combines maximal fragment assembly with chemical shift threading, de novo structure generation, chemical shift-based torsion angle prediction, and chemical shift refinement. CS23D makes use of RefDB and ShiftX.

CS23D input formats
CS23D accepts chemical shift files in either SHIFTY or BMRB formats.
CS23D options
A user can
- Exclude a protein from being used as the template
- Ignore high-identity homologs in the list of available templates
- Change the number of models in the final ensemble
- Change the number of model optimization steps
CS23D output
CS23D output consists of a set of 10 best-score PDB coordinates. A hyperlink to the single best score structure is also provided. The overall CS23D score, knowledge-based score, chemical shift score, Ramachandran plot statistics, correlations between the observed and calculated shifts before and after refinement are displayed. A conclusion about structure reliability is given to the user.
CS23D protocol
Homology search: The query sequence is used to find homologous proteins or/and protein fragments in a non-redundant database of PDB sequences and secondary structures of PPT-DB using BLAST.
Homology modelling: Homology modelling is done by the Homodeller program, which is a part of the PROTEUS2 program.[2] The proteins that are identified during the homology search step are used as the templates in homology modelling.
Chemical shift re-referencing: Chemical shifts are re-referenced by the RefCor,[3] which is a part of the RCI webserver backend.
Secondary structure prediction from chemical shifts: Secondary structure is predicted from chemical shifts by CSI.
Torsion angle prediction from chemical shifts: Torsion angles are predicted from chemical shifts by PREDITOR.[4]
Chemical shift threading: Backbone Phi and Psi torsion angles predicted from chemical shifts by PREDITOR[4] are mapped into nine different regions in Ramachandran space, each of which are assigned specific letters. A protein can represented by a sequence of these nine "torsion angle letters". Thrifty is using these sequences of torsion angle letters to identify good templates in a database of ~18,500 nonredundant PDB structures that have had their structures converted to the nine-letter Ramachandran "alphabet".
In a similar manner, chemical shift threading is additionally done using three-letter secondary structure alphabet (H for helix, B for beta-strand, C for coil) and secondary structure predicted from chemical shifts by the CSI program.
Model assembly: Subfragments identified by homology modelling and chemical shift threading steps are assembled into initial 3D models using CS23D SFassembler (SubFragment assembler). The initial models are evaluated by the GAFolder scoring function (see below) and the best model is further refined by GAFolder (see more info about GAFolder below).
Ab initio folding: Ab initio folding is done by Rosetta[5] when no template was identified by the homology modelling and chemical shift threading steps. Rosetta models are evaluated by GAFolder scoring function and the best Rosetta models are refined by GAFolder (see below).
Model optimization: Model optimization in CS23D is done by a torsion-angle-based minimizer GAfolder (Genetic Algorithm folder) that uses a genetic algorithm to sample conformation space. The method is similar to that employed by GENFOLD.[5] GAFolder makes torsion angles moves within the ranges defined by the values and uncertainties of torsion angles predicted by PREDITOR.[4] GAFolder evaluates protein models by the scoring function described below.
Scoring function: Scoring function of GAFolder consists of knowledge based scores and chemical shift scores.
The knowledge-based scores include:
- radius of gyration score,
- hydrogen bond energy,
- number of hydrogen bonds,
- bad contacts score,
- disulfide bond score,
- modified threading energy based on the Bryant and Lawrence potential.[6]
- Ramachandran score that evaluates normality of model torsion angles Phi and Psi
- Omega score that evaluates normality of model torsion omega angles
- Chi score that is based on expected chi angles for different phi and psi combinations.
The chemical shift component of the GAfolder scoring function uses:
- weighted coefficients of correlation between the experimental chemical shifts (CA, CB, CO, N, HA, HN) and chemical shifts calculated by SHIFTX 1.0.
- agreement between model secondary structure and secondary structure predicted by CSI from experimental chemical shifts.
CS23D sub-programs
- CSI - prediction of secondary structure from chemical shifts
- BLAST - sequence alignment, homology search
- PROTEUS2 - homology modelling[2]
- PREDITOR - prediction of torsion angles from chemical shifts[4]
- Pepmake - building protein models from torsion angles and sequence
- PPT-DB- secondary structure database
- Rosetta - ab initio structure generation[5]
- RCI- estimating uncertainty of torsion angles predicted from chemical shifts by PREDITOR
- ShiftX 1.0 - is used to generate coefficients of correlation between observed chemical shifts and shifts predicted by ShiftX from protein models
- SFAssembler - maximal fragment assembly
- GAFolder - chemical shift refinement via a genetic algorithm
- Thrifty - chemical shift threading
- RefCor - chemical shift re-referencing
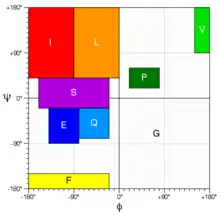
CS23D dependence on template sequence identity
CS23D is a template-based method. Therefore, its performance depends on sequence identity of the selected template(s), see the adjacent picture. Likewise, Rosetta is a fragment-biased method. Its performance depends on the quality of selected fragments. Fragment quality and, thus, Rosetta performance can be improved by using chemical shifts during the fragment selection step (e.g. in CS-Rosetta protocol). For a structural solution that is not biased by a template structure or fragment structure, one may want to consider obtaining NOE-based distance restraints (8-10 per residue) and using them with the GeNMR program in its ab initio mode.
See also
- Chemical Shift
- NMR
- Nuclear magnetic resonance spectroscopy
- Protein nuclear magnetic resonance spectroscopy
- Protein dynamics#Domains and protein flexibility
- Protein
- GeNMR
- Random Coil Index
- Protein Chemical Shift Prediction
- Protein Chemical Shift Re-Referencing
- Protein secondary structure
- Chemical shift index
- ShiftX
- Protein structure prediction
References
- Wishart DS, Arndt D, Berjanskii M, Tang P, Zhou J, Lin G (July 2008). "CS23D: a web server for rapid protein structure generation using NMR chemical shifts and sequence data". Nucleic Acids Research. 36 (Web Server issue): W496–502. doi:10.1093/nar/gkn305. PMC 2447725. PMID 18515350.
- Montgomerie S, Cruz JA, Shrivastava S, Arndt D, Berjanskii M, Wishart DS (July 2008). "PROTEUS2: a web server for comprehensive protein structure prediction and structure-based annotation". Nucleic Acids Research. 36 (Web Server issue): W202–9. doi:10.1093/nar/gkn255. PMC 2447806. PMID 18483082.
- Berjanskii M, Wishart DS (2006). "NMR: prediction of protein flexibility". Nature Protocols. 1 (2): 683–8. doi:10.1038/nprot.2006.108. PMID 17406296.
- Berjanskii MV, Neal S, Wishart DS (July 2006). "PREDITOR: a web server for predicting protein torsion angle restraints". Nucleic Acids Research. 34 (Web Server issue): W63–9. doi:10.1093/nar/gkl341. PMC 1538894. PMID 16845087.
- Rohl CA, Strauss CE, Misura KM, Baker D (2004). "Protein structure prediction using Rosetta". Numerical Computer Methods, Part D. Methods in Enzymology. Vol. 383. pp. 66–93. doi:10.1016/S0076-6879(04)83004-0. ISBN 9780121827885. PMID 15063647.
- Bryant SH, Lawrence CE (May 1993). "An empirical energy function for threading protein sequence through the folding motif". Proteins. 16 (1): 92–112. doi:10.1002/prot.340160110. PMID 8497488.