CUT&RUN sequencing
CUT&RUN sequencing, also known as cleavage under targets and release using nuclease, is a method used to analyze protein interactions with DNA. CUT&RUN sequencing combines antibody-targeted controlled cleavage by micrococcal nuclease with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global DNA binding sites precisely for any protein of interest. Currently, ChIP-Seq is the most common technique utilized to study protein–DNA relations, however, it suffers from a number of practical and economical limitations that CUT&RUN sequencing does not.
Uses
CUT&RUN sequencing can be used to examine gene regulation or to analyze transcription factor and other chromatin-associated protein binding. Protein-DNA interactions regulate gene expression and are responsible for many biological processes and disease states. This epigenetic information is complementary to genotype and expression analysis. CUT&RUN is an alternative to the current standard of ChIP-seq. ChIP-Seq suffers from limitations due to the cross linking step in ChIP-Seq protocols that can promote epitope masking and generate false-positive binding sites.[1][2] As well, ChIP-seq suffers from suboptimal signal-to-noise ratios and poor resolution.[3] CUT&RUN sequencing has the advantage of being a simpler technique with lower costs due to the high signal-to-noise ratio, requiring less depth in sequencing.[4]
Specific DNA sites in direct physical interaction with proteins such as transcription factors can be isolated by Protein-A (pA) conjugated micrococcal nuclease (MNase) bound to a protein of interest. MNase mediated cleavage produces a library of target DNA sites bound to a protein of interest in situ. Sequencing of prepared DNA libraries and comparison to whole-genome sequence databases allows researchers to analyze the interactions between target proteins and DNA, as well as differences in epigenetic chromatin modifications. Therefore, the CUT&RUN method may be applied to proteins and modifications, including transcription factors, polymerases, structural proteins, protein modifications, and DNA modifications.
Workflow
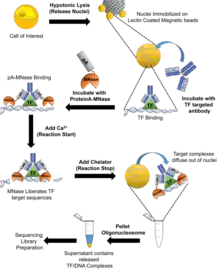
CUT&RUN is an adaptation and improvement on chromatin endogenous cleavage (ChEC) which uses a DNA-binding protein genetically fused to micrococcal nuclease (MNase). These transcription factor-MNase fusion proteins can cleave DNA around the DNA-binding site of the protein of interest.[5] In the adapted process, purified MNase is tagged with Protein A (pA) which targets an antibody that has been added to the cell and is specific for the DNA-binding protein that is of interest. There are seven general steps to the CUT&RUN process.
Cleavage under targets and release using nuclease
The first step required is the hypotonic lysis of the cells of interest to isolate the nuclei. The nuclei are then centrifuged, washed in a buffer solution, complexed with lectin-coated magnetic beads. The Lectin-Nuclei complex is then resuspended with an antibody targeted at the protein of interest. The antibody and nuclei are then incubated in the buffer for approximately 2 hours before the nuclei are washed in buffer to remove unbound antibodies. Next, the nuclei are resuspended in the buffer with Protein-A-MNase and are incubated for 1 hour. The nuclei are then again washed in buffer to remove any unbound protein-A-MNase. Next, the nuclei in tubes are placed in a metal block and placed in ice-water and CaCl2 is added to initiate the calcium dependent nuclease activity of MNase to cleave the DNA around the DNA-binding protein. The protein-A-MNase reaction is quenched by adding chelating agents (EDTA and EGTA). The cleaved DNA fragments are then liberated into the supernatant by incubating the nuclei for an hour before the nuclei is pelleted by centrifugation. The DNA fragments are then extracted from the supernatant and can be used to construct a sequencing library.
Sequencing
Unlike ChIP-Seq there is no size selection required before sequencing. A single sequencing run can scan for genome-wide associations with high resolution, due to the low background achieved by performing the reaction in situ with the CUT&RUN sequencing methodology. ChIP-Seq, by contrast, requires ten times the sequencing depth because of the intrinsically high background associated with the method.[6] The data is then collected and analyzed using software that aligns sample sequences to a known genomic sequence to identify the CUT&RUN DNA fragments.[4]
Protocols
There are detailed CUT&RUN workflows available in an open-access methods repository.
- CUT&RUN: Targeted in situ genome-wide profiling with high efficiency for low cell numbers[7]
- CUT&RUN with Drosophila tissues[8]
- AutoCUT&RUN: genome-wide profiling of chromatin proteins in a 96 well format on a Biomek[9]
- Bench top CUT&RUN with antibodies-online CUT&RUN Sets[10]
- CUT&RUN low volume-urea (LoV-U) for transcriptional co-factors[11]
Sensitivity
CUT&RUN sequencing provides low levels of background signal because of in situ profiling which retains in vivo 3D confirmations of transcription factor-DNA interactions, so antibodies access only exposed surfaces. Sensitivity of sequencing depends on the depth of the sequencing run (i.e. the number of mapped sequence tags), the size of the genome and the distribution of the target factor. The sequencing depth is directly correlated with cost and negatively correlated with background. Therefore, low-background CUT&RUN sequencing is inherently more cost-effective than high-background ChIP-Sequencing.
.tif.png.webp)
Current research
There have already been a number of research projects that have made use of the new CUT&RUN technology.
In humans, researchers looking at fetal globin gene promoters have used CUT&RUN to investigate the involvement of the protein BCL11A in mediating the function of the HBBP1 gene region,[12][13] highlighting a potential target for therapeutic genome editing for hemoglobinopathies.
A research group has used CUT&RUN to identify intermediates involved in nucleosome disruption during DNA transcription,[14] validating a general strategy for structural epigenomics.
In humans and in African green monkeys, researchers using CUT&RUN determined that the CENP-B protein (an important protein in centromere formation) and binding sites are specific to great ape centromeres,[15] addressing the paradox that CENP-B, which is required for artificial centromere function, is non-essential.
Computational analysis
As with many high-throughput sequencing approaches, CUT&RUN-seq generates extremely large data sets, for which appropriate computational analysis methods are required. To predict DNA-binding sites from CUT&RUN-seq read count data, peak calling methods have been developed.
Peak calling is a process where an algorithm is used to predict the regions of the genome that a transcription factor binds to by finding regions of the genome that have many mapped reads from a ChIP-seq or CUT&RUN-seq experiment. MACS is a particularly popular peak calling algorithm for ChIP-seq data.[16] SEACR is a highly selective peak caller that definitively validates the accuracy of CUT&RUN for datasets with known true negatives.[17]
To identify the causal DNA-binding motif for CUT&RUN-seq peak calls one can apply the MEME motif-finding program to the CUT&RUN sequences. This involves using a position-specific scoring matrix (PSSM) along with the Motif Alignment and Search Tool (MAST) to identify motifs in a reference genome that match the acquired sequence reads.[4] This process allows the identification of the transcription-factor binding motif, or if the binding motif was previously known, this process can act to confirm the success of the experiment[18]
Limitations
The primary limitation of CUT&RUN-seq is the likelihood of over-digestion of DNA due to inappropriate timing of the Calcium-dependent MNase reaction. A similar limitation exists for contemporary ChIP-Seq protocols where enzymatic or sonicated DNA shearing must be optimized. As with ChIP-Seq, a good quality antibody targeting the protein of interest is required.
Similar methods
- Sono-Seq: Identical to ChIP-Seq but without the immunoprecipitation step.
- HITS-CLIP: Also called CLIP-Seq, employed to detect interactions with RNA rather than DNA.
- PAR-CLIP: A method for identifying the binding sites of cellular RNA-binding proteins.
- RIP-Chip: Similar to ChIP-Seq, but does not employ cross linking methods and utilizes microarray analysis instead of sequencing.
- SELEX: Employed to determine consensus binding sequences.
- Competition-ChIP: Measures relative replacement dynamics on DNA.
- ChiRP-Seq: Measures RNA-bound DNA and proteins.
- ChIP-exo: Employs exonuclease treatment to achieve up to single base-pair resolution
- ChIP-nexus: Potential improvement on ChIP-exo, capable of achieving up to single base-pair resolution.
- DRIP-seq: Employs S9.6 antibody to precipitate three-stranded DND:RNA hybrids called R-loops.
- TCP-seq: Principally similar method to measure mRNA translation dynamics.
- DamID: Uses enrichment of methylated DNA sequences to detect protein-DNA interaction without antibodies.
See also
References
- Meyer CA, Liu XS (November 2014). "Identifying and mitigating bias in next-generation sequencing methods for chromatin biology". Nature Reviews. Genetics. 15 (11): 709–21. doi:10.1038/nrg3788. PMC 4473780. PMID 25223782.
- Baranello L, Kouzine F, Sanford S, Levens D (May 2016). "ChIP bias as a function of cross-linking time". Chromosome Research. 24 (2): 175–81. doi:10.1007/s10577-015-9509-1. PMC 4860130. PMID 26685864.
- He C, Bonasio R (February 2017). "A cut above". eLife. 6. doi:10.7554/eLife.25000. PMC 5310838. PMID 28199181.
- Skene PJ, Henikoff S (January 2017). "An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites". eLife. 6. doi:10.7554/eLife.21856. PMC 5310842. PMID 28079019.
- "Lay off the ChIPs: CUT&RUN instead". Fred Hutchinson Cancer Research Center. 20 February 2017.
- "Still Using ChIP? Try CUT&RUN for Enhanced Chromatin Profiling". EpiCypher. Retrieved 2019-07-26.
- Janssens, Derek; Henikoff, Steven (9 May 2019). "CUT&RUN: Targeted in situ genome-wide profiling with high efficiency for low cell numbers v3 (protocols.io.zcpf2vn)". protocols.io. doi:10.17504/protocols.io.zcpf2vn.
- Ahmad, Kami (2 December 2018). "CUT&RUN with Drosophila tissues v1 (protocols.io.umfeu3n)". protocols.io. doi:10.17504/protocols.io.umfeu3n. S2CID 216840246.
- Janssens, Derek; Ahmad, Kami; Henikoff, Steven (27 November 2018). "AutoCUT&RUN: genome-wide profiling of chromatin proteins in a 96 well format on a Biomek v1 (protocols.io.ufeetje)". protocols.io. doi:10.17504/protocols.io.ufeetje. S2CID 216872521.
- antibodies-online (19 March 2020). "Bench top CUT&RUN with antibodies-online CUT&RUN Sets (protocols.io.bdwni7de)". protocols.io. doi:10.17504/protocols.io.bdwni7de.
- Zambanini, Gianluca; Nordin, Anna; Jonasson, Mattias; Pagella, Pierfrancesco; Cantù, Claudio (1 December 2022). "A new CUT&RUN low volume-urea (LoV-U) protocol optimized for transcriptional co-factors uncovers Wnt/β-catenin tissue-specific genomic targets". Development. 149 (23). doi:10.1242/dev.201124. PMID 36355069. S2CID 253445603.
- Huang P, Keller CA, Giardine B, Grevet JD, Davies JO, Hughes JR, Kurita R, Nakamura Y, Hardison RC, Blobel GA (August 2017). "Comparative analysis of three-dimensional chromosomal architecture identifies a novel fetal hemoglobin regulatory element". Genes & Development. 31 (16): 1704–1713. doi:10.1101/gad.303461.117. PMC 5647940. PMID 28916711.
- Liu N, Hargreaves VV, Zhu Q, Kurland JV, Hong J, Kim W, Sher F, Macias-Trevino C, Rogers JM, Kurita R, Nakamura Y, Yuan GC, Bauer DE, Xu J, Bulyk ML, Orkin SH (April 2018). "Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch". Cell. 173 (2): 430–442.e17. doi:10.1016/j.cell.2018.03.016. PMC 5889339. PMID 29606353.
- Ramachandran S, Ahmad K, Henikoff S (December 2017). "Transcription and Remodeling Produce Asymmetrically Unwrapped Nucleosomal Intermediates". Molecular Cell. 68 (6): 1038–1053.e4. doi:10.1016/j.molcel.2017.11.015. PMC 6421108. PMID 29225036.
- Kasinathan S, Henikoff S (April 2017). "Non-B-Form DNA Is Enriched at Centromeres". Molecular Biology and Evolution. 35 (4): 949–962. doi:10.1093/molbev/msy010. PMC 5889037. PMID 29365169.
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS (2008). "Model-based analysis of ChIP-Seq (MACS)". Genome Biology. 9 (9): R137. doi:10.1186/gb-2008-9-9-r137. PMC 2592715. PMID 18798982.
- Meers MP, Tenenbaum D, Henikoff S (July 2019). "Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling". Epigenetics & Chromatin. 12 (1): 42. doi:10.1186/s13072-019-0287-4. PMC 6624997. PMID 31300027.
- Bailey T, Krajewski P, Ladunga I, Lefebvre C, Li Q, Liu T, Madrigal P, Taslim C, Zhang J (2013). "Practical guidelines for the comprehensive analysis of ChIP-seq data". PLOS Computational Biology. 9 (11): e1003326. Bibcode:2013PLSCB...9E3326B. doi:10.1371/journal.pcbi.1003326. PMC 3828144. PMID 24244136.