Circulating tumor DNA
Circulating tumor DNA (ctDNA) is tumor-derived fragmented DNA in the bloodstream that is not associated with cells. ctDNA should not be confused with cell-free DNA (cfDNA), a broader term which describes DNA that is freely circulating in the bloodstream, but is not necessarily of tumor origin. Because ctDNA may reflect the entire tumor genome, it has gained traction for its potential clinical utility; "liquid biopsies" in the form of blood draws may be taken at various time points to monitor tumor progression throughout the treatment regimen.[1][2]
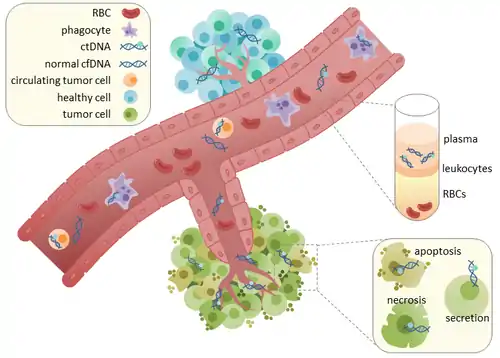
ctDNA originates directly from the tumor or from circulating tumor cells (CTCs),[3] which describes viable, intact tumor cells that shed from primary tumors and enter the bloodstream or lymphatic system. The precise mechanism of ctDNA release is unclear. The biological processes postulated to be involved in ctDNA release include apoptosis and necrosis from dying cells, or active release from viable tumor cells.[4][5][6][7][8] Studies in both human (healthy and cancer patients)[9] and xenografted mice[10] show that the size of fragmented cfDNA is predominantly 166bp long, which corresponds to the length of DNA wrapped around a nucleosome plus a linker. Fragmentation of this length might be indicative of apoptotic DNA fragmentation, suggesting that apoptosis may be the primary method of ctDNA release. The fragmentation of cfDNA is altered in the plasma of cancer patients.[11][12] In healthy tissue, infiltrating phagocytes are responsible for clearance of apoptotic or necrotic cellular debris, which includes cfDNA.[13] ctDNA in healthy patients is only present at low levels but higher levels of ctDNA in cancer patients can be detected with increasing tumor sizes.[14] This possibly occurs due to inefficient immune cell infiltration to tumor sites, which reduces effective clearance of ctDNA from the bloodstream.[13] Comparison of mutations in ctDNA and DNA extracted from primary tumors of the same patients revealed the presence of identical cancer-relevant genetic changes.[15][16] This led to the possibility of using ctDNA for earlier cancer detection and treatment follow up.[17]
Methods
Pre-analytical considerations
When blood is collected in EDTA tubes and stored, the white blood cells begin to lyse and release genomic wild type DNA in to the sample in quantities typically many fold higher than the ctDNA is present in.[18] This makes detection of mutations or other ctDNA biomarkers more difficult.[19] The use of commercially available cell stabilisation tubes can prevent or delay the lysis of white cells thereby reducing the dilution effect of the ctDNA.[20] Sherwood et al. demonstrated superior detection of KRAS mutations in matched samples collected in both EDTA K3 and Streck BCT tubes.[20] The advantages of cell stabilisation tubes can be realised in situation where blood cannot be processed to plasma immediately.
Other procedures can also reduce the amount of "contaminating" wild type DNA and make detection of ctDNA more feasible:[20]
- Never freeze a blood sample before extracting the plasma for ctDNA analysis
- Process the sample to plasma within 2–4 hours (if collected in EDTA tube)
- Never use heparinised tubes, heparin inhibits PCR by mimicking the helical structure of DNA
- Perform a double centrifugation step (centrifuge the blood to extract plasma, then repeat on the plasma to remove from debris in the bottom of the tube) to remove more cellular debris prior to DNA extraction.
- Plasma is better than serum for ctDNA recovery[21]
Extraction of ctDNA
The main appeal of ctDNA analysis is that it is extracted in a non-invasive manner through blood collection. Acquisition of cfDNA or ctDNA typically requires collection of approximately 3mL of blood into EDTA-coated tubes. The use of EDTA is important to reduce coagulation of blood. The plasma and serum fractions of blood can be separated through a centrifugation step. ctDNA or cfDNA can be subsequently extracted from these fractions. Although serum tends to have greater levels of cfDNA, this is primarily attributed to DNA from lymphocytes.[22] High levels of contaminating cfDNA is sub-optimal because this can decrease the sensitivity of ctDNA detection. Therefore, the majority of studies use plasma for ctDNA isolation. Plasma is then processed again by centrifugation to remove residual intact blood cells. The supernatant is used for DNA extraction, which can be performed using commercially available kits.
Analysis of ctDNA
The analysis of ctDNA after extraction requires the use of various amplification and sequencing methods. These methods can be separated into two main groups based on whether the goal is to interrogate all genes in an untargeted approach, or if the goal is to monitor specific genes and mutations in a targeted approach.
Untargeted approaches
A whole genome or whole exome sequencing approaches may be necessary to discover new mutations in tumor DNA while monitoring disease burden or tracking drug resistance.[23] Untargeted approaches are also useful in research to observe tumor heterogeneity or to discover new drug targets. However, while untargeted methods may be necessary in certain applications, it is more expensive and has lower resolution. This makes it difficult to detect rare mutations, or in situations where low ctDNA levels are present (such as minimal residual disease). Furthermore, there can be problems distinguishing between DNA from tumor cells and DNA from normal cells using a whole genome approach.
Whole genome or exome sequencing typically use high throughput DNA sequencing technologies. Limiting the sequencing to only the whole exome instead can decrease expense and increase speed, but at the cost of losing information about mutations in the non-coding regulatory regions of DNA.[24] While simply looking at DNA polymorphisms through sequencing does not differentiate DNA from tumor or normal cells, this problem can be resolved by comparing against a control sample of normal DNA (for example, DNA obtained through a buccal swab.) Importantly, whole genome and whole exome sequencing are useful for initial mutation discovery. This provides information for the use of more sensitive targeted techniques, which can then be used for disease monitoring purposes.
Whole genome sequencing enables to recover the structural properties of cfDNA, the size of fragments and their fragmentation patterns. These unique patterns can be an important source of information to improve the detection of ctDNA or localize the tissue of origin of these fragments.[25] Size-selection of short fragments (<150bp) with in vitro or in silico methods could improve the recovery of mutations and copy number aberrations.[12]
Digital Karyotyping
This method was originally developed by the laboratory of Bert Vogelstein, Luis Diaz, and Victor Velculescu at Johns Hopkins University.[26] Unlike normal karyotyping where a dye is used to stain chromosomal bands in order to visualize the chromosomes, digital karyotyping uses DNA sequences of loci throughout the genome in order to calculate copy number variation.[26] Copy number variations are common in cancers and describe situations where loss of heterozygosity of a gene may lead to decreased function due to lower expression, or duplication of a gene, which leads to overexpression.
Personalized analysis of rearranged ends (PARE)
After the whole genome is sequenced using a high throughput sequencing method, such as Illumina HiSeq, PARE is applied to the data to analyze chromosomal rearrangements and translocations. This technique was originally designed to analyze solid tumor DNA but was modified for ctDNA applications.[26]
DNA Methylation and Hydroxymethylation
Proper epigenetic marking is essential for normal gene expression and cell function and aberrant alterations in epigenetic patterns is a hallmark of cancer.[27] A normal epigenetic status is maintained in a cell at least in part through DNA methylation.[28] Measuring aberrant methylation patterns in ctDNA is possible due to stable methylation of regions of DNA referred to as “CpG islands”. Methylation of ctDNA can be detected through bisulfite treatment. Bisulfite treatment chemically converts unmethylated cytosines into a uracil while leaving methylated cytosines unmodified. DNA is subsequently sequenced, and any alterations to the DNA methylation pattern can be identified. DNA hydroxymethylation is a similarly associated mark that has been shown to be a predictive marker of healthy versus diseased conditions in cfDNA, including cancer.[29][30])
Targeted approaches
In a targeted approach, sequencing of ctDNA can be directed towards a genetic panel constructed based on mutational hotspots for the cancer of interest. This is especially important for informing treatment in situations where mutations are identified in druggable targets.[24] Personalizing targeted analysis of ctDNA to each patient is also possible by combining liquid biopsies with standard primary tissue biopsies. Whole genome or whole exome sequencing of the primary tumor biopsy allows for discovery of genetic mutations specific to a patient's tumor, and can be used for subsequent targeted sequencing of the patient's ctDNA. The highest sensitivity of ctDNA detection is accomplished through targeted sequencing of specific single nucleotide polymorphisms (SNPs). Commonly mutated genes, such as oncogenes, which typically have hotspot mutations, are good candidates for targeted sequencing approaches. Conversely, most tumor suppressor genes have a wide array of possible loss of function mutations throughout the gene, and as such are not suitable for targeted sequencing.
Targeted approaches have the advantage of amplifying ctDNA through polymerase chain reactions (PCR) or digital PCR. This is especially important when analyzing ctDNA not only because there are relatively low levels of DNA circulating in the bloodstream, but also because ctDNA makes up a small proportion of the total cell-free DNA extracted.[24] Therefore, amplification of regions of interest can drastically improve sensitivity of ctDNA detection. However, amplification through PCR can introduce errors given the inherent error rate of DNA polymerases. Errors introduced during sequencing can also decrease the sensitivity of detecting ctDNA mutations.
Droplet Digital PCR (ddPCR)
This method is derived from the digital PCR, originally named by Bert Vogelstein’s group at Johns Hopkins University. Droplet Digital PCR utilizes a droplet generator to partition single pieces of DNA into droplets using an oil/water emulsion. Then individual polymerase chain reactions occur in each droplet using selected primers against regions of ctDNA and proceeds to endpoint. The presence of the sequences of interest is measured by fluorescent probes, which bind to the amplified region. ddPCR allows for highly quantitative assessment of allele and mutant frequencies in ctDNA but is limited by the number of fluorescent probes that can be used in one assay (up to 5).[31] The sensitivity of the assay can vary depending on the amount of DNA analyzed and is around 1 in 10,000.[31] Specificity should be augmented through the use of either minor groove binding (MGB) modified probes or of an alternative such as Locked Nucleic Acids (LNA's).
Beads, Emulsification, Amplification, and Magnetics (BEAMing)
This technique builds upon Droplet Digital PCR in order to identify mutations in ctDNA using flow cytometry.[32] After ctDNA is extracted from blood, PCR is performed with primers designed to target the regions of interest. These primers also contain specific DNA sequences, or tags. The amplified DNA is mixed with streptavidin-coated magnetic beads and emulsified into droplets. Biotinylated primers designed to bind to the tags are used to amplify the DNA. Biotinylation allows the amplified DNA to bind to the magnetic beads, which are coated with streptavidin. After the PCR is complete, the DNA-bound beads are separated using a magnet. The DNA on the beads are then denatured and allowed to hybridize with fluorescent oligonucleotides specific to each DNA template. The resulting bead-DNA complexes are then analyzed using flow cytometry. This technique is able to capture allele and mutation frequencies due to coupling with ddPCR. However, unlike with ddPCR, a larger number of DNA sequences can be interrogated due to the flexibility of using fluorescently bound probes. Another advantage of this system is that the DNA isolated can also be used for downstream sequencing.[33] Sensitivity is 1.6 in 104 to 4.3 in 105.[31]
Cancer Personalized Profiling by deep Sequencing (CAPP-Seq)
This method was originally described by Ash Alizadeh and Maximilian Diehn's groups at Stanford University. This technique uses biotinylated oligonucleotide selector probes to target sequences of DNA relevant to ctDNA detection.[34] Publicly available cancer databases were used to construct a library of probes against recurrent mutations in cancer by calculating their recurrence index. The protocol was optimized for the low DNA levels observed in ctDNA collection. Then the isolated DNA undergoes deep sequencing for increased sensitivity. This technique allows for the interrogation of hundreds of DNA regions. The ctDNA detection sensitivity of CAPP-Seq is reported to be 2.5 molecules in 1,000,000.[35]
Tagged AMplicon deep Sequencing (TAM-Seq)
TAM-Seq allows targeted sequencing of entire genes to detect mutations in ctDNA.[36] First a general amplification step is performed using primers that span the entire gene of interest in 150-200bp sections. Then, a microfluidics system is used to attached adaptors with a unique identifier to each amplicon to further amplify the DNA in parallel singleplex reactions. This technique was shown to successfully identify mutations scattered in the TP53 tumor suppressor gene in advanced ovarian cancer patients. The sensitivity of this technique is 1 in 50.
Safe-Sequencing (Safe-Seq)
This method was originally described by Bert Vogelstein and his group at Johns Hopkins University. Safe-Seq decreases the error rate of massively parallel sequencing in order to increase the sensitivity to rare mutants.[37] It achieves this by addition of a unique identifier (UID) sequence to each DNA template. The DNA is then amplified using the added UIDs and sequenced. All DNA molecules with the same UID (a UID family) should have the same reported DNA sequence since they were amplified from one molecule. However, mutations can be introduced through amplification, or incorrect base assignments may be called in the sequencing and analysis steps. The presence of the UID allows these methodology errors to be separated from true mutations of the ctDNA. A mutation is considered a ‘supermutant’ if 95% of the sequenced reads are in agreement. The sensitivity of this approach is 9 in 1 million.[31]
Duplex sequencing
This method is an improvement on the single UIDs added in the Safe-Seq technique.[38] In duplex sequencing, randomized double-stranded DNA act as unique tags and are attached to an invariant spacer. Tags are attached to both ends of a DNA fragment (α and β tags), which results in two unique templates for PCR - one strand with an α tag on the 5’ end and a β tag on the 3’ end and the other strand with a β tag on the 5’ end and an α tag on the 3’ end. These DNA fragments are then amplified with primers against the invariant sequences of the tags. The amplified DNA is sequenced and analyzed. DNA with the duplex adaptors are compared and mutations are only accepted if there is a consensus between both strands. This method takes into account both errors from sequencing and errors from early stage PCR amplification. The sensitivity of the approach to discovering mutants is 1 in 10^7.
Integrated Digital Error Suppression (iDES)-enhanced CAPP-Seq
iDES improves CAPP-Seq analysis of ctDNA in order to decrease error and therefore increase sensitivity of detection.[35] Reported in 2016, iDES combines CAPP-Seq with duplex barcoding sequencing technology and with a computational algorithm that removes stereotypical errors associated with the CAPP-Seq hybridization step. The method also integrates duplex sequencing where possible, and includes methods for more efficient duplex recovery from cell free DNA. The sensitivity of this improved version of CAPP-Seq is 4 in 100,000 copies.
Whole-genome sequencing
Whole-genome sequencing investigations have been performed on ctDNA present in different patients with treatment-resistant prostate cancer (the vast majority, and in some cases, metastatic), bladder cancer and control patients who did not present this DNA, including somatic mutations and structural rearrangements in their genomes.
This novel and promising technique has provided information on resistance to treatment with androgen receptor signaling inhibitors, intratumoral heterogeneity (thanks to phylogenetic evolution and molecular chronology), chromosomal instability, contribution of ctDNA to metastasis through global transcriptomic patterns (taking into account nucleosomes present in transcription start sites (TSSs) and AR-binding sites (ARBs). In this way, the genomic and transcriptomic evolution of ctDNA can be observed, performed in living patients who are developing resistance to treatment, therefore, ctDNA sequencing is elementary to identify clinically relevant differences in the cancer phenotype and to see how therapy is affecting patients. Furthermore, the relative homogeneity in driver gene alterations among metastases justifies that genomic and functional alterations in prostate cancer are shared between ctDNA and tissue.
This makes ctDNA a powerful emerging tool for the detection of genetic mutations at the genomic scale in patients suffering from metastatic cancer to observe the clinical relevance of the clonal composition of these tumors to understand better cancer control. This subclonal reconstruction based on ctDNA thanks to Whole-genome sequencing poses a unique set of challenges and opportunities for scientific research in oncology. Furthermore, serial ctDNA reveals treatment-driven selection for androgen receptor augmentation because it increases the dimensionality of the data.
Further work is needed to understand how metastatic location and size, in relation to tumor burden, influence circulating tumor DNA and the choice of new techniques to select other lesions that reflect clinically dominant disease. [39]
Considerations
“Normal” vs tumor DNA detection
One of the challenges in using ctDNA as a cancer biomarker is whether ctDNA can be distinguished with cfDNA from normal cells. cfDNA is released by non-malignant cells during normal cellular turnover, but also during procedures such as surgery, radiotherapy, or chemotherapy. It is thought that leukocytes are the primary contributors to cfDNA in serum.[24]
Research
ctDNA in cancer screening
The clinical utility of ctDNA for the detection of primary disease is in part limited by the sensitivity of current technology to detect small tumors with low levels of ctDNA present and a priori unknown somatic mutations.[14][31]
ctDNA in cancer monitoring
Evidence of disease by traditional imaging methods, such as CT, PET or MRI may be absent after tumor resection. Therefore, ctDNA analysis poses a potential avenue to detect minimal residual disease (MRD), and thus the possibility of tumor recurrence, in cases where bulk tumors are absent by conventional imaging methods.[14] A comparison of MRD detection by CT imaging compared to ctDNA has been previously done in individuals with stage II colon cancer; in this study, researchers were able to detect ctDNA in individuals who showed no sign of clinical malignancy by a CT scan, suggesting that ctDNA detection has greater sensitivity to assess MRD.[24] However, the authors acknowledge that ctDNA analysis is not without limitations; plasma samples collected post-operatively were only able to predict recurrence at 36 months in 48% of cases.[24] Subsequently, ctDNA assays have been developed for both colorectal cancer[40] and melanoma.[41]
ctDNA as a prognostic biomarker
The question of whether measurement of the amount or qualities of ctDNA could be used to determine outcomes in people with cancer has been a subject of study. As of 2015 this was very uncertain.[42] Although some studies have shown a trend of higher ctDNA levels in people with high stage metastatic cancer, ctDNA burden does not always correlate with traditional cancer staging.[31] As of 2013 it appeared unlikely that ctDNA would be of clinical utility as a sole predictor of prognosis.[43]
Cancer research
The emergence of drug-resistant tumors due to intra- and inter-tumoral heterogeneity an issue in treatment efficacy. A minor genetic clone within the tumor can expand after treatment if it carries a drug-resistant mutation. Initial biopsies can miss these clones due to low frequency or spatial separation of cells within the tumor. For example, since a biopsy only samples a small part of the tumor, clones that resides in a different location may go unnoticed. This can mislead research that focuses on studying the role of tumor heterogeneity in cancer progression and relapse. The use of ctDNA in research can alleviate these concerns because it could provide a more representative 'screenshot' of the genetic diversity of cancer at both primary and metastatic sites. For example, ctDNA has been shown to be useful in studying the clonal evolution of a patient's cancer before and after treatment regimens.[44] Early detection of cancer is still challenging but recent progress in the analysis of the epigenetic features of cfDNA, or the fragmentation pattern unlock improve the sensitivity of liquid biopsy.[25] Furthermore, ctDNA analysis is an emerging tool for understanding the clonal composition of metastatic tumors, detecting different mutations on a genomic scale, studying subclonal diversity that affects the prognosis of the disease as different resistant phenotypes can be found and the appearance of new mechanisms of genomic and transcriptomic resistance to treatment.[39]
Challenges for implementation
Implementation of ctDNA in clinical practice is largely hindered by the lack of standardized methods for ctDNA processing and analysis. Standardization of methods for sample collection (including time of collection), downstream processing (DNA extraction and amplification), quantification and validation must be established before ctDNA analysis can become a routine clinical assay. Furthermore, creation of a panel of ‘standard’ tumor-associated biomarkers may be necessary given the resolution of current ctDNA sequencing and detection methods. Sequencing tumor-specific aberrations from plasma samples may also help exclude contaminating cfDNA from analysis; elevated levels of cfDNA from normal cells may be attributed to non-cancer related causes.[24] These sequencing techniques can also determine the clonal evolution of cancer, tumor heterogeneity and drug resistance mechanisms involved in cancer.[39]
See also
References
- Wan J, Massie C, Garcia-Corbacho J, Mouliere F, Brenton J, Caldas C, Pacey S, Baird R, Rosenfeld N (April 2017). "Liquid biopsies come of age: towards implementation of circulating tumour DNA". Nature Reviews Cancer. 17 (4): 223–238. doi:10.1038/nrc.2017.7. PMID 28233803. S2CID 4561229.
- Nonaka, T; Wong, DTW (13 June 2022). "Saliva Diagnostics". Annual Review of Analytical Chemistry. 15 (1): 107–121. Bibcode:2022ARAC...15..107N. doi:10.1146/annurev-anchem-061020-123959. PMC 9348814. PMID 35696523.
- Akca H, Demiray A, Yaren A, Bir F, Koseler A, Iwakawa R, Bagci G, Yokota J (March 2013). "Utility of serum DNA and pyrosequencing for the detection of EGFR mutations in non-small cell lung cancer". Cancer Genetics. 206 (3): 73–80. doi:10.1016/j.cancergen.2013.01.005. PMID 23491080.
- Schwarzenbach H, Hoon DS, Pantel K (June 2011). "Cell-free nucleic acids as biomarkers in cancer patients". Nature Reviews. Cancer. 11 (6): 426–37. doi:10.1038/nrc3066. PMID 21562580. S2CID 6061607.
- Stroun M, Anker P (July 1972). "Nucleic acids spontaneously released by living frog auricles". The Biochemical Journal. 128 (3): 100P–101P. doi:10.1042/bj1280100pb. PMC 1173871. PMID 4634816.
- Stroun M, Lyautey J, Lederrey C, Olson-Sand A, Anker P (November 2001). "About the possible origin and mechanism of circulating DNA apoptosis and active DNA release". Clinica Chimica Acta; International Journal of Clinical Chemistry. 313 (1–2): 139–42. doi:10.1016/S0009-8981(01)00665-9. PMID 11694251.
- Anker P, Stroun M, Maurice PA (September 1975). "Spontaneous release of DNA by human blood lymphocytes as shown in an in vitro system". Cancer Research. 35 (9): 2375–82. PMID 1149042.
- Rogers JC, Boldt D, Kornfeld S, Skinner A, Valeri CR (July 1972). "Excretion of deoxyribonucleic acid by lymphocytes stimulated with phytohemagglutinin or antigen". Proceedings of the National Academy of Sciences of the United States of America. 69 (7): 1685–9. Bibcode:1972PNAS...69.1685R. doi:10.1073/pnas.69.7.1685. PMC 426778. PMID 4505646.
- Heitzer E, Auer M, Hoffmann EM, Pichler M, Gasch C, Ulz P, Lax S, Waldispuehl-Geigl J, Mauermann O, Mohan S, Pristauz G, Lackner C, Höfler G, Eisner F, Petru E, Sill H, Samonigg H, Pantel K, Riethdorf S, Bauernhofer T, Geigl JB, Speicher MR (July 2013). "Establishment of tumor-specific copy number alterations from plasma DNA of patients with cancer". International Journal of Cancer. 133 (2): 346–56. doi:10.1002/ijc.28030. PMC 3708119. PMID 23319339.
- Thierry AR, Mouliere F, Gongora C, Ollier J, Robert B, Ychou M, Del Rio M, Molina F (October 2010). "Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts". Nucleic Acids Research. 38 (18): 6159–75. doi:10.1093/nar/gkq421. PMC 2952865. PMID 20494973.
- Mouliere F, Robert B, Arnau Peyrotte E, Del Rio M, Ychou M, Molina F, Gongora C, Thierry AR (2011). "High fragmentation characterizes tumour-derived circulating DNA". PLOS ONE. 6 (9): e23418. Bibcode:2011PLoSO...623418M. doi:10.1371/journal.pone.0023418. PMC 3167805. PMID 21909401.
- Mouliere F, Chandrananda D, Piskorz AM, Moore EK, Morris J, Ahlborn LB, et al. (November 2018). "Enhanced detection of circulating tumor DNA by fragment size analysis". Sci Transl Med. 10 (466). doi:10.1126/scitranslmed.aat4921. PMC 6483061. PMID 30404863.
- Pisetsky DS, Fairhurst AM (June 2007). "The origin of extracellular DNA during the clearance of dead and dying cells". Autoimmunity. 40 (4): 281–4. doi:10.1080/08916930701358826. PMID 17516210. S2CID 11499768.
- Avanzini S, Kurtz DM, Chabon JJ, Moding EJ, Hori SS, Gambhir SS, Alizadeh AA, Diehn M, Reiter JG (December 2020). "A mathematical model of ctDNA shedding predicts tumor detection size". Science Advances. 6 (50): eabc4308. Bibcode:2020SciA....6.4308A. doi:10.1126/sciadv.abc4308. PMC 7732186. PMID 33310847. S2CID 228096858.
- Vasioukhin V, Anker P, Maurice P, Lyautey J, Lederrey C, Stroun M (April 1994). "Point mutations of the N-ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia". British Journal of Haematology. 86 (4): 774–779. doi:10.1111/j.1365-2141.1994.tb04828.x. PMID 7918071. S2CID 26365875.
- Vasioukhin V, Stroun M, Maurice P, Lyautey J, Lederrey C, Anker P (May 1994). K-ras point mutations in the blood plasma DNA of patients with colorectal tumors in Challenges of Modern Medicine. Vol. 5. pp. 141–150.
- Yong E (July 2014). "Cancer biomarkers: Written in blood". Nature. 511 (7511): 524–526. Bibcode:2014Natur.511..524Y. doi:10.1038/511524a. PMID 25079538. S2CID 4445938.
- Xue X, Teare MD, Holen I, Zhu YM, Woll PJ (June 2009). "Optimizing the yield and utility of circulating cell-free DNA from plasma and serum" (PDF). Clinica Chimica Acta; International Journal of Clinical Chemistry. 404 (2): 100–4. doi:10.1016/j.cca.2009.02.018. PMID 19281804.
- Norton SE, Lechner JM, Williams T, Fernando MR (October 2013). "A stabilizing reagent prevents cell-free DNA contamination by cellular DNA in plasma during blood sample storage and shipping as determined by digital PCR". Clinical Biochemistry. 46 (15): 1561–5. doi:10.1016/j.clinbiochem.2013.06.002. PMID 23769817.
- Sherwood JL, Corcoran C, Brown H, Sharpe AD, Musilova M, Kohlmann A (2016). "Optimised Pre-Analytical Methods Improve KRAS Mutation Detection in Circulating Tumour DNA (ctDNA) from Patients with Non-Small Cell Lung Cancer (NSCLC)". PLOS ONE. 11 (2): e0150197. Bibcode:2016PLoSO..1150197S. doi:10.1371/journal.pone.0150197. PMC 4769175. PMID 26918901.
- Vallée A, Marcq M, Bizieux A, Kouri CE, Lacroix H, Bennouna J, Douillard JY, Denis MG (November 2013). "Plasma is a better source of tumor-derived circulating cell-free DNA than serum for the detection of EGFR alterations in lung tumor patients". Lung Cancer. 82 (2): 373–4. doi:10.1016/j.lungcan.2013.08.014. PMID 24007628.
- Lee TH, Montalvo L, Chrebtow V, Busch MP (February 2001). "Quantitation of genomic DNA in plasma and serum samples: higher concentrations of genomic DNA found in serum than in plasma". Transfusion. 41 (2): 276–82. doi:10.1046/j.1537-2995.2001.41020276.x. PMID 11239235. S2CID 45714834.
- Qin Z, Ljubimov VA, Zhou C, Tong Y, Liang J (April 2016). "Cell-free circulating tumor DNA in cancer". Chinese Journal of Cancer. 35: 36. doi:10.1186/s40880-016-0092-4. PMC 4823888. PMID 27056366.
- Heitzer E, Ulz P, Geigl JB (January 2015). "Circulating tumor DNA as a liquid biopsy for cancer". Clinical Chemistry. 61 (1): 112–23. doi:10.1373/clinchem.2014.222679. PMID 25388429.
- van der Pol Y, Mouliere F (October 2019). "Toward the Early Detection of Cancer by Decoding the Epigenetic and Environmental Fingerprints of Cell-Free DNA". Cancer Cell. 36 (4): 350–368. doi:10.1016/j.ccell.2019.09.003. PMID 31614115.
- Leary RJ, Sausen M, Kinde I, Papadopoulos N, Carpten JD, Craig D, O'Shaughnessy J, Kinzler KW, Parmigiani G, Vogelstein B, Diaz LA, Velculescu VE (November 2012). "Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing". Science Translational Medicine. 4 (162): 162ra154. doi:10.1126/scitranslmed.3004742. PMC 3641759. PMID 23197571.
- Preobrazhenskiĭ BS (1966). "[Current perspectives and the method of systematic treatment of cochlear neuritis and chronic labyrinthopathy]". Vestnik Otorinolaringologii (in Russian). 28 (1): 3–11. PMID 5988180.
- Beaumont G, Dobbins S, Latta D, McMillin WP (May 1990). "Mequitazine in the treatment of hayfever". The British Journal of Clinical Practice. 44 (5): 183–8. PMID 1975200.
- Li W, Zhang X, Lu X, You L, Song Y, Luo Z, et al. (October 2017). "5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers". Cell Research. 27 (10): 1243–1257. doi:10.1038/cr.2017.121. PMC 5630683. PMID 28925386.
- Song CX, Yin S, Ma L, Wheeler A, Chen Y, Zhang Y, Liu B, Xiong J, Zhang W, Hu J, Zhou Z, Dong B, Tian Z, Jeffrey SS, Chua MS, So S, Li W, Wei Y, Diao J, Xie D, Quake SR (October 2017). "5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages". Cell Research. 27 (10): 1231–1242. doi:10.1038/cr.2017.106. PMC 5630676. PMID 28820176.
- Butler TM, Spellman PT, Gray J (February 2017). "Circulating-tumor DNA as an early detection and diagnostic tool". Current Opinion in Genetics & Development. 42: 14–21. doi:10.1016/j.gde.2016.12.003. PMID 28126649.
- Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B (July 2003). "Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations". Proceedings of the National Academy of Sciences of the United States of America. 100 (15): 8817–22. Bibcode:2003PNAS..100.8817D. doi:10.1073/pnas.1133470100. PMC 166396. PMID 12857956.
- Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D (July 2006). "BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions". Nature Methods. 3 (7): 551–9. doi:10.1038/nmeth898. PMID 16791214. S2CID 7059151.
- Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, Liu CL, Neal JW, Wakelee HA, Merritt RE, Shrager JB, Loo BW, Alizadeh AA, Diehn M (May 2014). "An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage". Nature Medicine. 20 (5): 548–54. doi:10.1038/nm.3519. PMC 4016134. PMID 24705333.
- Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. (May 2016). "Integrated digital error suppression for improved detection of circulating tumor DNA". Nature Biotechnology. 34 (5): 547–555. doi:10.1038/nbt.3520. PMC 4907374. PMID 27018799.
- Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, Dawson SJ, Piskorz AM, Jimenez-Linan M, Bentley D, Hadfield J, May AP, Caldas C, Brenton JD, Rosenfeld N (May 2012). "Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA". Science Translational Medicine. 4 (136): 136ra68. doi:10.1126/scitranslmed.3003726. PMID 22649089. S2CID 34723244.
- Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B (June 2011). "Detection and quantification of rare mutations with massively parallel sequencing". Proceedings of the National Academy of Sciences of the United States of America. 108 (23): 9530–5. Bibcode:2011PNAS..108.9530K. doi:10.1073/pnas.1105422108. PMC 3111315. PMID 21586637.
- Kennedy SR, Schmitt MW, Fox EJ, Kohrn BF, Salk JJ, Ahn EH, Prindle MJ, Kuong KJ, Shen JC, Risques RA, Loeb LA (November 2014). "Detecting ultralow-frequency mutations by Duplex Sequencing". Nature Protocols. 9 (11): 2586–606. doi:10.1038/nprot.2014.170. PMC 4271547. PMID 25299156.
- Herberts, Cameron; Annala, Matti; Sipola, Joonatan; Ng, Sarah W. S.; Chen, Xinyi E.; Nurminen, Anssi; Korhonen, Olga V.; Munzur, Aslı D.; Beja, Kevin; Schönlau, Elena; Bernales, Cecily Q.; Ritch, Elie; Bacon, Jack V. W.; Lack, Nathan A.; Nykter, Matti (August 2022). "Deep whole-genome ctDNA chronology of treatment-resistant prostate cancer". Nature. 608 (7921): 199–208. Bibcode:2022Natur.608..199H. doi:10.1038/s41586-022-04975-9. ISSN 1476-4687. PMID 35859180. S2CID 250730778.
- Zou, Donghui; Day, Robert; Cocadiz, Judy A; Parackal, Sarah; Mitchell, Wilson; Black, Michael A; Lawrence, Ben; Fitzgerald, Sandra; Print, Cristin; Jackson, Christopher; Guilford, Parry (2020-11-13). "Circulating tumor DNA is a sensitive marker for routine monitoring of treatment response in advanced colorectal cancer". Carcinogenesis. 41 (11): 1507–1517. doi:10.1093/carcin/bgaa102. ISSN 0143-3334. PMID 32955091.
- Fitzgerald, Sandra; Blenkiron, Cherie; Stephens, Rosalie; Mathy, Jon A.; Somers-Edgar, Tiffany; Rolfe, Gill; Martin, Richard; Jackson, Christopher; Eccles, Michael; Robb, Tamsin; Rodger, Euan; Lawrence, Ben; Guilford, Parry; Lasham, Annette; Print, Cristin G. (2023). "Dynamic ctDNA Mutational Complexity in Patients with Melanoma Receiving Immunotherapy". Molecular Diagnosis & Therapy. 27 (4): 537–550. doi:10.1007/s40291-023-00651-4. ISSN 1177-1062. PMC 10131510. PMID 37099071.
- Rapisuwon S, Vietsch EE, Wellstein A (2016). "Circulating biomarkers to monitor cancer progression and treatment". Computational and Structural Biotechnology Journal. 14: 211–22. doi:10.1016/j.csbj.2016.05.004. PMC 4913179. PMID 27358717.
- Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A (August 2013). "Liquid biopsy: monitoring cancer-genetics in the blood". Nature Reviews. Clinical Oncology. 10 (8): 472–84. doi:10.1038/nrclinonc.2013.110. PMID 23836314. S2CID 25537784.
- Murtaza M, Dawson SJ, Pogrebniak K, Rueda OM, Provenzano E, Grant J, Chin SF, Tsui DW, Marass F, Gale D, Ali HR, Shah P, Contente-Cuomo T, Farahani H, Shumansky K, Kingsbury Z, Humphray S, Bentley D, Shah SP, Wallis M, Rosenfeld N, Caldas C (November 2015). "Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer". Nature Communications. 6 (1): 8760. Bibcode:2015NatCo...6.8760M. doi:10.1038/ncomms9760. PMC 4659935. PMID 26530965.
Further reading
- Applying circulating tumor DNA methylation in the diagnosis of lung cancer May 2019
- Circulating tumor DNA: A new generation of cancer biomarkers Feb 2014
- ctDNA 'Liquid Biopsy' Could Revolutionize Cancer Care Nov 2014
- Karachaliou N, Mayo-de-Las-Casas C, Molina-Vila MA, Rosell R (March 2015). "Real-time liquid biopsies become a reality in cancer treatment". Annals of Translational Medicine. 3 (3): 36. doi:10.3978/j.issn.2305-5839.2015.01.16. PMC 4356857. PMID 25815297.
- Marusina, Kate (8 February 2018). "Teasing Out Circulating Tumor DNA". ClinicalOMICs. Retrieved 5 March 2018.
- Du-Bois, Asante (2019). "Liquid biopsy in ovarian cancer using circulating tumor DNA and cells: Ready for prime time?". Cancer Letters. 468: 59–71. doi:10.1016/j.canlet.2019.10.014. PMID 31610267.