ChIA-PET
Chromatin Interaction Analysis by Paired-End Tag Sequencing (ChIA-PET or ChIA-PETS) is a technique that incorporates chromatin immunoprecipitation (ChIP)-based enrichment, chromatin proximity ligation, Paired-End Tags, and High-throughput sequencing to determine de novo long-range chromatin interactions genome-wide.[1]
Genes can be regulated by regions far from the promoter such as regulatory elements, insulators and boundary elements, and transcription-factor binding sites (TFBS). Uncovering the interplay between regulatory regions and gene coding regions is essential for understanding the mechanisms governing gene regulation in health and disease (Maston et al., 2006). ChIA-PET can be used to identify unique, functional chromatin interactions between distal and proximal regulatory transcription-factor binding sites and the promoters of the genes they interact with.
ChIA-PET can also be used to unravel the mechanisms of genome control during processes such as cell differentiation, proliferation, and development. By creating ChIA-PET interactome maps for DNA-binding regulatory proteins and promoter regions, we can better identify unique targets for therapeutic intervention (Fullwood & Yijun, 2009).
Methodology
The ChIA-PET method combines ChIP-based methods,[2] and Chromosome conformation capture (3C) based methods,[3] to extend the capabilities of both approaches. ChIP-Sequencing (ChIP-Seq) is a popular method used to identify TFBS while 3C has been used to identify long-range chromatin interactions. Independently, both suffer from limitations in identifying de-novo long-range interactions genome wide. While ChIP-Seq is able to identify TFBS genome-wide,[4][5] it provides only linear information of protein binding sites along the chromosomes (but not interactions between them), and can suffer from high genomic background noise (false positives). While 3C is capable of analyzing non-linear, long-range chromatin interactions, it cannot be used genome wide and, like ChIP-Seq, also suffers from high levels of background noise. Since the noise increases in relation to the distance between interacting regions (max 100kb), laborious and tedious controls are required for accurate characterization of chromatin interactions.[6] Unlike 3C which is a locus-specific interaction profiling method, alternative methods such as Hi-C have been established to profile interactions genome wide.[7] Despite whole genome profiling methods for both TFBS and long range interactions, combining approaches with the ChIA-PET method allows for identification of genomic areas in which the protein of interest is bound as well as the genomic region which it interacts with.[8][9]
The ChIA-PET method successfully resolves the issues of non-specific interaction noise found in ChIP-Seq by sonicating the ChIP fragments in order to separate random attachments from specific interaction complexes. The next step, which is referred to as enrichment, reduces complexity for genome-wide analysis and adds specificity to chromatin interactions bound by pre-determined TFs (transcription factors). The ability of 3C approaches to identify long-range interactions is based on the theory of proximity ligation. In regards to DNA inter-ligation, fragments that are tethered by common protein complexes have greater kinetic advantages under dilute conditions, than those freely diffusing in solution or anchored in different complexes. ChIA-PET takes advantage of this concept by incorporating linker sequences onto the free ends of the DNA fragments tethered to the protein complexes. In order to build connectivity of the fragments tethered by regulatory complexes, the linker sequences are ligated during nuclear proximity ligation. Therefore, the products of linker-connected ligation can be analyzed by ultra-high-throughput PET sequencing and mapped to the reference genome. Since ChIA-PET is not dependent on specific sites for detection as 3C and 4C are, it allows unbiased, genome-wide de-novo detection of chromatin interactions.[8] Compared to Hi-C, the use of an antibody pulldown limits the number of sequenced fragments to chromatin interactions bound by the protein of interest which also can ease the data analysis.
Workflow
Wet-lab portion of the workflow:

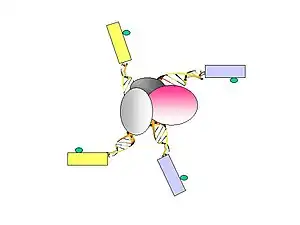
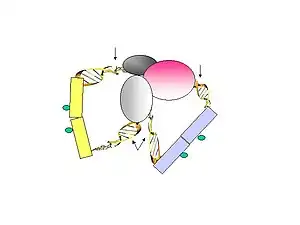
- Formaldehyde is used to cross-link the DNA-protein complexes. Sonication is used to break-up the chromatin and also to reduce non-specific interactions.
- A specific antibody of choice is used to enrich protein-of-interest–bound chromatin fragments. ChIP material bound by the antibody are used to construct the ChIA-PET.
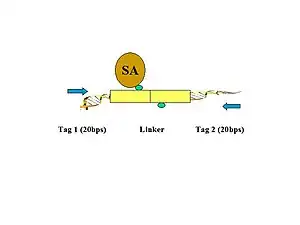
- Figure 1. Biotinylated oligonucleotide half-linkers containing flanking MmeI sites are used to connect proximity ligated DNA fragments. Two different linkers are designed (A and B) with specific nucleotide barcodes (CG or AT) for each of the two linker sequences (this will allow the identification of the chimeric ligation product as described in Figure 5.).
- Figure 2. The linkers are ligated to the tethered DNA fragments.
- Figure 3. The linker fragments are ligated on the ChIP beads under dilute conditions. The purified DNA is then digested by MmeI, which cuts at a distance from its recognition site to release the tag-linker-tag structure.
- Figure 4. The biotinylated PETs are then immobilized on streptavidin-conjugated magnetic beads.
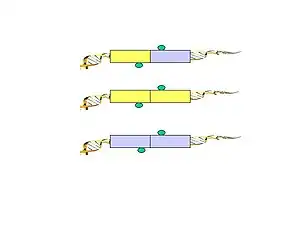
- Figure 5. PET sequences with AA (CG/CG) and BB (AT/AT) linker barcode composition are considered to be possible intra-complex ligation products, while the PET sequences with AB (CG/AT) linker composition are considered to be derived from chimeric ligation products between DNA fragments bounded in different chromatin complexes.
Dry-lab portion of the workflow:
PET extraction, mapping, and statistical analyses
The PET tags are extracted and mapped to the reference human genome in silico.
Identification of ChIP enriched peaks (binding sites)
Self-ligated PET are used for identifying ChIP enriched sites because they provide the most reliable mapping (20 + 20 bit/s) to the reference genome.
ChIP enrichment peak-finding algorithm
A called peak is considered a binding site if there are multiple overlapping self-ligated PETs. The false discovery rate (FDR) is determined using statistical simulations to estimate the random background of PET-derived virtual DNA overlaps, and the estimated background noise.
Filtering of repetitive DNA (affects non-specific binding)
Satellite regions and binding sites present in regions with severe structural variations are removed.
ChIP enrichment count
The numbers of self-ligation and inter-ligation PETs (within + 250 bp window) are reported at each site. The total number of self-ligated and inter-ligated PETs at a specific site is called the ChIP enrichment count.
Figure 6. PET Classification: Uniquely aligned PET sequences can be classified by whether they are derived from one DNA fragment or two DNA fragments.
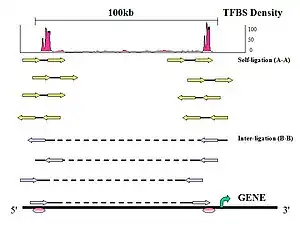
- Self-ligation PETs
If the two tags of a PET are mapped on the same chromosome with the genomic span in the range of ChIP DNA fragments (less than 3 Kb), with expected self-ligation orientation and on the same strand, they are considered to be derived from a self-ligation of a single ChIP DNA fragment, and considered a self-ligation PET.
- Inter-ligation PETs
If a PET does not fit into these criteria, then the PET most likely resulted from a ligation product between two DNA fragments and referred to as an inter-ligation PET. The two tags of an inter-ligation PETs do not have fixed tag orientations, might not be found on the same strands, might have any genomic span, and might not map to the same chromosome.
- Intrachromosomal inter-ligation PETs
If the two tags of an inter-ligation PET are mapped in the same chromosome but with a span > 3 Kb in any orientation, then these PETs are called intrachromosomal inter-ligation PETs.
- Interchromosomal inter-ligation PETs
PETs which are mapped to different chromosomes are called interchromosomal inter-ligation PETs.
Figure 7. Proposed mechanism showing how distal regulatory elements can initiate long-range chromatin interactions involving promoter regions of target genes.
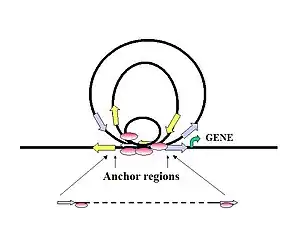
The interactions form DNA loop structures with multiple TFBS at the anchoring center. Small loops might package genes near the anchoring center in a tight sub-compartment, which could increase the local concentration of regulatory proteins for enhanced transcriptional activation. This mechanism might also enhance transcription efficiency, allowing RNA pol II to cycle the tight circular gene templates. The large interaction loops are more likely to link together distant genes at either end of the loop residing near anchor sites for coordinated regulation, or could separate genes in long loops to prevent their activation. Adapted from Fullwood et al. (2009).
Strengths and weaknesses
Advantages of the ChIA-PET method
- ChIA-PET has a potential to be an unbiased, whole-genome and de-novo approach for long-range chromatin interaction analysis (Fullwood & Yijun, 2009).
- A ChIA-PET experiment is capable of providing two global datasets: The protein factor binding sites (self-ligated PETs); and the interactions between the binding sites (inter-ligated PETs).
- ChIA-PET involves ChIP to reduce the complexity for genome-wide analysis and adds specificity to chromatin interactions bound by specific factors of interest.
- ChIA-PET is compatible with tag-based next-generation sequencing approaches such as Roche 454 pyrosequencing, Illumina GA, ABI SOLiD, and Helicos.
- ChIA-PET is applicable to many different protein factors involved in transcriptional regulation or chromatin structural conformation.
- ChIA-PET analysis can be applied to chromatin interactions involved in a particular nuclear process. By using general TFs such as RNA Polymerase II, it may be possible to identify all chromatin interactions involved in transcription regulation. Further, the use of protein factors involved in DNA replication or chromatin structure would allow identification of all interactions due to DNA replication and chromatin structural modification (Fullwood et al., 2009).
Weaknesses
- It is well established that cis and trans-regulatory complexes contain unique combinations of proteins based on cell and tissue specific conditions (Dekker et al., 2006). While identification of single, functional TFBS is a significant advancement, the use of ChIA-PET to identify individual proteins in a complex would require guess work and multiple experiments to identify each interacting protein. This would be a costly and time-consuming endeavour.
- ChIA-PET is limited by the quality, purity, and specificity of the antibodies used (Fullwood et al., 2009).
- ChIA-PET is dependent on identification of sequences that can be mapped to the reference sequence (ref).
- ChIA-PET requires the use of peak-calling computer algorithms to organize and map PET reads to the reference genome. Because of variations between software platforms, results can vary depending on which program is used.
- Although repetitive DNA regions can be associated with gene regulation (Polak & Domany, 2006), they need to be removed as they can affect the data (Fullwood et al., 2009).
- Enrichment from two sites simultaneously introduces bias against surrounding regions.[10]
History
Fullwood et al. (2009), used ChIA-PET to detect and map the chromatin interaction network mediated by estrogen receptor alpha (ER-alpha) in human cancer cells. The resulting global chromatin interactome map revealed that remote ER-alpha-binding sites were also anchored to gene promoters through long-range chromatin interactions suggesting that ER-alpha functions by extensive chromatin looping in order to bring genes together for coordinated transcriptional regulation.
Analysis and software
| Software | Description |
|---|---|
| C3PET | A software suite for processing ChIA-PET data. Uses a non-parametric Bayesian approach to predict chromatin interacting protein complexes. |
| ChIA-PET Tool | A software suite for processing ChIA-PET data. |
| ChIA-PET2 | A software suite for processing ChIA-PET data. Supports data from a variety of protocols and provides quality control of data analysis. |
| ChIA-Sig | A software suite for processing ChIA-PET data using the NCHG model. ChIA-Sig web site |
| ELAND | Maps ChIP enriched DNA fragments to the reference human genome. |
| Mango | A software suite for processing ChIA-PET data. Completes all required steps of processing ChIA-PET datasets and provides statistical confidence estimates for interactions. |
| Monte Carlo Simulation | Used to estimate the false discovery rates.[11] |
| GenomicInteractions | An R package for processing ChIA-PET or Hi-C data. |
| GIVE | A programming library for creating a custom genome browser compatible with ChIA-PET or Hi-C data. |
| RepeatMasker | In-silico masking of repetitive elements. |
Alternatives
Chromatin immunoprecipitation (ChIP):

The original ChIP method is an antibody-based technology that identify and bind proteins selectively in order to offer information regarding chromatin states and gene transcription.
Genome Architecture Mapping (GAM):
This technique eliminates a number of drawbacks associated with 3C-based techniques by collecting three-dimensional proximities between any number of genomic loci.[12]
Split-Pool Recognition of Interactions by Tag Extension (SPRITE)
SPRITE is a technique for mapping higher-order interactions in the nucleus across the genome. This approach detects interactions that occur over greater spatial distances and it allows for genome-wide detection of numerous RNA and DNA interactions that occur at the same time.[13]
References
- Fullwood, Melissa J.; Ruan, Yijun (2009-05-01). "ChIP-based methods for the identification of long-range chromatin interactions". Journal of Cellular Biochemistry. 107 (1): 30–39. doi:10.1002/jcb.22116. ISSN 1097-4644. PMC 2748757. PMID 19247990.
- Kuo, Min-Hao; Allis, C.David (November 1999). "In Vivo Cross-Linking and Immunoprecipitation for Studying Dynamic Protein:DNA Associations in a Chromatin Environment". Methods. 19 (3): 425–433. doi:10.1006/meth.1999.0879. PMID 10579938.
- Dekker, Job; Rippe, Karsten; Dekker, Martijn; Kleckner, Nancy (2002-02-15). "Capturing Chromosome Conformation". Science. 295 (5558): 1306–1311. Bibcode:2002Sci...295.1306D. doi:10.1126/science.1067799. ISSN 0036-8075. PMID 11847345. S2CID 3561891.
- Barski, Artem; Cuddapah, Suresh; Cui, Kairong; Roh, Tae-Young; Schones, Dustin E.; Wang, Zhibin; Wei, Gang; Chepelev, Iouri; Zhao, Keji (May 2007). "High-Resolution Profiling of Histone Methylations in the Human Genome". Cell. 129 (4): 823–837. doi:10.1016/j.cell.2007.05.009. PMID 17512414. S2CID 6326093.
- Wei, Chia-Lin; Wu, Qiang; Vega, Vinsensius B.; Chiu, Kuo Ping; Ng, Patrick; Zhang, Tao; Shahab, Atif; Yong, How Choong; Fu, YuTao; Weng, Zhiping; Liu, JianJun (January 2006). "A Global Map of p53 Transcription-Factor Binding Sites in the Human Genome". Cell. 124 (1): 207–219. doi:10.1016/j.cell.2005.10.043. PMID 16413492. S2CID 15698887.
- Dekker, Job (January 2006). "The three 'C' s of chromosome conformation capture: controls, controls, controls". Nature Methods. 3 (1): 17–21. doi:10.1038/nmeth823. ISSN 1548-7091. PMID 16369547. S2CID 25821556.
- Lieberman-Aiden, Erez; van Berkum, Nynke L.; Williams, Louise; Imakaev, Maxim; Ragoczy, Tobias; Telling, Agnes; Amit, Ido; Lajoie, Bryan R.; Sabo, Peter J.; Dorschner, Michael O.; Sandstrom, Richard (2009-10-09). "Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome". Science. 326 (5950): 289–293. Bibcode:2009Sci...326..289L. doi:10.1126/science.1181369. ISSN 0036-8075. PMC 2858594. PMID 19815776.
- Fullwood, Melissa J.; Liu, Mei Hui; Pan, You Fu; Liu, Jun; Xu, Han; Mohamed, Yusoff Bin; Orlov, Yuriy L.; Velkov, Stoyan; Ho, Andrea; Mei, Poh Huay; Chew, Elaine G. Y. (November 2009). "An oestrogen-receptor-α-bound human chromatin interactome". Nature. 462 (7269): 58–64. Bibcode:2009Natur.462...58F. doi:10.1038/nature08497. ISSN 0028-0836. PMC 2774924. PMID 19890323.
- Li, Guoliang; Ruan, Xiaoan; Auerbach, Raymond K.; Sandhu, Kuljeet Singh; Zheng, Meizhen; Wang, Ping; Poh, Huay Mei; Goh, Yufen; Lim, Joanne; Zhang, Jingyao; Sim, Hui Shan (2012-01-20). "Extensive Promoter-Centered Chromatin Interactions Provide a Topological Basis for Transcription Regulation". Cell. 148 (1): 84–98. doi:10.1016/j.cell.2011.12.014. ISSN 0092-8674. PMC 3339270. PMID 22265404.
- Downes, Damien J.; Beagrie, Robert A.; Gosden, Matthew E.; Telenius, Jelena; Carpenter, Stephanie J.; Nussbaum, Lea; De Ornellas, Sara; Sergeant, Martin; Eijsbouts, Chris Q.; Schwessinger, Ron; Kerry, Jon (2021-01-22). "High-resolution targeted 3C interrogation of cis-regulatory element organization at genome-wide scale". Nature Communications. 12 (1): 531. Bibcode:2021NatCo..12..531D. doi:10.1038/s41467-020-20809-6. ISSN 2041-1723. PMC 7822813. PMID 33483495.
- Monte Carlo method
- Beagrie, Robert A.; Scialdone, Antonio; Schueler, Markus; Kraemer, Dorothee C. A.; Chotalia, Mita; Xie, Sheila Q.; Barbieri, Mariano; de Santiago, Inês; Lavitas, Liron-Mark; Branco, Miguel R.; Fraser, James (March 2017). "Complex multi-enhancer contacts captured by genome architecture mapping". Nature. 543 (7646): 519–524. Bibcode:2017Natur.543..519B. doi:10.1038/nature21411. ISSN 1476-4687. PMC 5366070. PMID 28273065.
- Quinodoz, Sofia A.; Ollikainen, Noah; Tabak, Barbara; Palla, Ali; Schmidt, Jan Marten; Detmar, Elizabeth; Lai, Mason M.; Shishkin, Alexander A.; Bhat, Prashant; Takei, Yodai; Trinh, Vickie (2018-07-26). "Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus". Cell. 174 (3): 744–757.e24. doi:10.1016/j.cell.2018.05.024. ISSN 0092-8674. PMC 6548320. PMID 29887377. S2CID 47013513.
- Zheng, Meizhen; Tian, Simon Zhongyuan; Capurso, Daniel; Kim, Minji; Maurya, Rahul; Lee, Byoungkoo; Piecuch, Emaly; Gong, Liang; Zhu, Jacqueline Jufen; Li, Zhihui; Wong, Chee Hong (February 2019). "Multiplex chromatin interactions with single-molecule precision". Nature. 566 (7745): 558–562. Bibcode:2019Natur.566..558Z. doi:10.1038/s41586-019-0949-1. ISSN 1476-4687. PMC 7001875. PMID 30778195.
- Koch, Linda (April 2019). "Getting the drop on chromatin interaction". Nature Reviews Genetics. 20 (4): 192–193. doi:10.1038/s41576-019-0103-9. ISSN 1471-0064. PMID 30783222. S2CID 67752274.
- Barski et al., (2007). High-resolution profiling of histone methylations in the human genome. Cell. (129); 823–37.
- Dekker, (2002). Capturing chromosome conformation. Science. (295); 1306–1311.
- Dekker, (2006). The three ‘C’ s of chromosome conformation capture: controls, controls, controls. Nat. Methods. (3); 17–21.
- Fullwood et al., (2009). An oestrogen-receptor-α bound human chromatin interactome. Nature. (462); 58–64.
- Fullwood & Yijun, (2009). ChIP-based methods for the identification of long-range chromatin interactions. J Cell Biochem. 107(1); 30–39.
- Johnson et al., (2007). Genome-wide mapping of in vivo protein-DNA interactions. Science. (316); 1497–502.
- Kuo & Allis, (1999). In-vivo cross-linking and immunoprecipitation for studying dynamic Protein: DNA associations in a chromatin environment. Methods. (19); 425–33.
- Li, G., Fullwood, M.J., Xu, H., Mulawadi, F.H., Velkov, S., Vega, V., Ariyaratne, P.N., Mohamed, Y.B., Ooi, H.S., Tennakoon, C., Wei, C.L., Ruan, Y. and Sung, W.K. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol, 11 (2). R22.
- Maston et al., (2006). Transcriptional Regulatory Elements in the Human Genome. Annu. Rev: Genomics. Hum Genet. (7); 29–59.
- Polak & Domany, (2006). Alu elements contain many binding sites for transcription factors and may play a role in regulation of developmental processes. BMC Genomics. (7); 133.
- Wei et al., (2006). A global map of p53 transcription-factor binding sites in the human genome. Cell. (124); 207–19.
External links
- ChIA-PET Genome Browser - This browser is for viewing the data from Fullwood et al. (2009), and includes a custom Whole Genome Interaction Viewer which provides a macroscopic picture of binding sites and interactions along with a whole genome landscape.