Chemoproteomics
Chemoproteomics (also known as chemical proteomics) entails a broad array of techniques used to identify and interrogate protein-small molecule interactions.[1] Chemoproteomics complements phenotypic drug discovery, a paradigm that aims to discover lead compounds on the basis of alleviating a disease phenotype, as opposed to target-based drug discovery (reverse pharmacology), in which lead compounds are designed to interact with predetermined disease-driving biological targets.[2] As phenotypic drug discovery assays do not provide confirmation of a compound's mechanism of action, chemoproteomics provides valuable follow-up strategies to narrow down potential targets and eventually validate a molecule's mechanism of action.[2] Chemoproteomics also attempts to address the inherent challenge of drug promiscuity in small molecule drug discovery by analyzing protein-small molecule interactions on a proteome-wide scale. A major goal of chemoproteomics is to characterize the interactome of drug candidates to gain insight into mechanisms of off-target toxicity and polypharmacology.[3]
Chemoproteomics assays can be stratified into three basic types. Solution-based approaches involve the use of drug analogs that chemically modify target proteins in solution, tagging them for identification. Immobilization-based approaches seek to isolate potential targets or ligands by anchoring their binding partners to an immobile support. Derivatization-free approaches aim to infer drug-target interactions by observing changes in protein stability or drug chromatography upon binding. Computational techniques complement the chemoproteomic toolkit as parallel lines of evidence supporting potential drug-target pairs, and are used to generate structural models that inform lead optimization. Several targets of high profile drugs have been identified using chemoproteomics, and the continued improvement of mass spectrometer sensitivity and chemical probe technology indicates that chemoproteomics will play a large role in future drug discovery.
Background
Context
The conclusion of the Human Genome Project was followed with hope for a new paradigm in treating disease. Many fatal and intractable diseases were able to be mapped to specific genes, providing a starting point to better understand the roles of their protein products in illness. Drug discovery has made use of animal knock-out models that highlight the impact of a protein's absence, particularly in the development of disease, and medicinal chemists have leveraged computational chemistry to generate high affinity compounds against disease-causing proteins.[3] Yet FDA drug approval rates have been on the decline over the last decade.[4] One potential source of drug failure is the disconnect between early and late drug discovery.[3] Early drug discovery focuses on genetic validation of a target, which is a strong predictor of success, but knock-out and overexpression systems are simplistic. Spatially and temporally conditional knock-out/knock-in systems have improved the level of nuance in in vivo analysis of protein function, but still fail to completely parallel the systemic breadth of pharmacological action.[3] For example, drugs often act through multiple mechanisms, and often work best by engaging targets partially.[3] Chemoproteomic tools offer a solution to bridge the gap between a genetic understanding of disease and a pharmacological understanding of drug action by identifying the many proteins involved in therapeutic success.
Basic tools
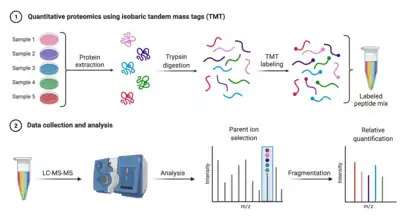
The chemoproteomic toolkit is anchored by liquid chromatography-tandem mass spectrometry (LC-MS/MS or LC-MS) based quantitative proteomics, which allows for the near complete identification and relative quantification of complex proteomes in biological samples.[5] In addition to proteomic analysis, the detection of post-translational modifications, like phosphorylation, glycosylation, acetylation, and recently ubiquitination, which give insight into the functional state of a cell, is also possible.[5] The vast majority of proteomic studies are analyzed using high-resolution orbitrap mass spectrometers and samples are processed using a generalizable workflow. A standard procedure begins with sample lysis, in which proteins are extracted into a denaturing buffer containing salts, an agent that reduces disulfide bonds, such as dithiothreitol, and an alkylating agent that caps thiol groups, such as iodoacetamide. Denatured proteins are proteolysed, often with trypsin, and then separated from other mixture components prior to analysis via LC-MS/MS. For more accurate quantification, different samples can be reacted with isobaric tandem mass tags (TMTs), a form of chemical barcode that allows for sample multiplexing, and then pooled.[5]
Solution-based approaches
Broad proteomic and transcriptomic profiling has led to innumerable advances in the biomedical space, but the characterization of RNA and protein expression is limited in its ability to inform on the functional characteristics of proteins. Given that transcript and protein expression information leave gaps in knowledge surrounding the effects of post-translational modifications and protein-protein interactions on enzyme activity, and that enzyme activity varies across cell types, disease states, and physiological conditions, specialized tools are required to profile enzyme activity across contexts.[6] Additionally, many identified enzymes have not been sufficiently characterized to yield actionable mechanisms on which to base functional assays. Without a basis for a functional biochemical readout, chemical tools are required to detect drug-protein interactions.
Activity-based protein profiling
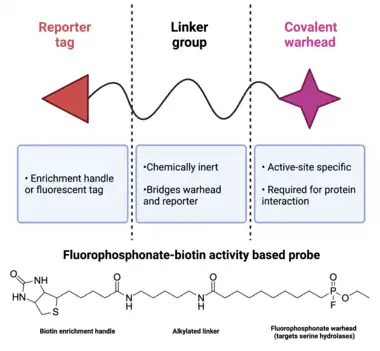
Activity-based protein profiling (ABPP, also activity-based proteomics) is a technique that was developed to monitor the availability of enzymatic active sites to their endogenous ligands.[5] ABPP uses specially designed probes that enter and form a covalent bond with an enzyme's active site, which confirms that the enzyme is an active state.[7] The probe is typically an analog of the drug whose mechanism is being studied, so covalent labeling of an enzyme is indicative of drug binding. ABPP probes are designed with three key functional units: (1) a site-directed covalent warhead (reactive group); (2) a reporter tag, such as biotin or rhodamine; and (3) a linker group.[8] The site-directed covalent warhead, also called a covalent modifier, is an electrophile that covalently modifies a serine, cysteine, or lysine residue in the enzyme's active site and prevents future interactions with other ligands.[7] ABPP probes are generally designed against enzymatic classes, and thus can provide systems-level information about the impact of cell state on enzymatic networks. The reporter tag is used to confirm labeling of the enzyme with the reactive group and can vary depending on the downstream readout. The most widely used reporters are fluorescent moieties that enable imaging and affinity tags, such as biotin, that allow for pull-down of labeled enzymes and analysis via mass spectrometry. There are drawbacks to each strategy, namely that fluorescent reporters do not allow for enrichment for proteomic analysis, while biotin-based affinity tags co-purify with endogenously biotinylated proteins.[8] A linker group is used to connect the reactive group to the reporter, ideally in a manner that does not alter the activity of probe. The most common linker groups are long alkyl chains, derivatized PEGs, and modified polypeptides.[5]
Under the assumption that enzymes vary in their structure, function, and associations depending on a system's physiological or developmental state, it can be inferred that the accessibility of an enzyme's active site will also vary.[6] Therefore, the ability of an ABPP probe to label an enzyme will also vary across conditions. Thus, the binding of a probe can reveal information around an enzyme's functional characteristics in different contexts. High-throughput screening has benefitted from ABPP, particularly in the area of competitive inhibition assays, in which biological samples are pre-incubated with drug candidates, then made to compete with ABPP probes for binding to target enzymes. Compounds with high affinity to their targets will prevent binding of the probe, and the degree of probe binding can be used as an indication of compound affinity. Because ABPP probes label classes of enzymes, this approach can also be used to profile drug selectivity, as highly selective compounds will ideally outcompete probes at only a small number of proteins.[8]
Photoaffinity labeling
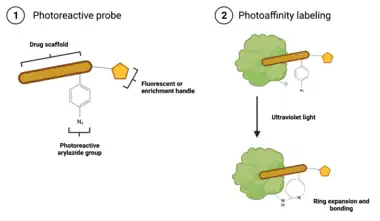
Unlike ABPP, which results in protein labeling upon probe binding, photoaffinity labeling probes require activation by photolysis before covalent bonding to a protein occurs.[7] The presence of a photoreactive group makes this possible.[7] These probes are composed of three connected moieties: (1) a drug scaffold; (2) a photoreactive group, such as an phenylazide, phenyldiazirine, or benzophenone; and (3) an identification tag, such as biotin, a fluorescent dye, or a click chemistry handle.[9] The drug scaffold is typically an analog of a drug whose mechanism is being studied, and, importantly, binds to the target reversibly, which better mimics the interaction between most drugs and their targets.[9] There are several varieties of photoreactive groups, but they are fundamentally different from ABPP probes: while ABPP specifically labels nucleophilic amino acids in a target's active site, photoaffinity labeling is non-specific, and thus is applicable to labeling a wider range of targets.[7] The identification tag will vary depending on the type of analysis being done: biotin and click chemistry handles are suitable for enrichment of labeled proteins prior to mass spectrometry based identification, while fluorescent dyes are used when using a gel-based imaging method, such as SDS-PAGE, to validate interaction with a target.[7][9]
Because photoaffinity probes are multifunctional, they are difficult to design. Chemists incorporate the same principles of structure-activity relationship modeling into photoaffinity probes that apply to drugs, but must do so without compromising the drug scaffold's activity or the photoreactive group's ability to bond.[9] Since photoreactive groups bond indiscriminately, improper design can cause the probe to label itself or non-target proteins. The probe must remain stable in storage, across buffers, at various pH levels, and in living systems to ensure that labeling occurs only when exposed to light. Activation by light must also be fine-tuned, as radiation can damage cells.[9]
Immobilization-based approaches
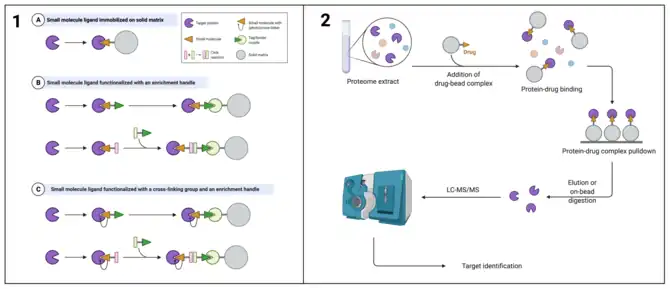
Immobilization-based chemoproteomic techniques encompass variations on microbead-based affinity pull-down, which is similar to immunoprecipitation, and affinity chromatography. In both cases, a solid support is used as an immobilization surface bearing a bait molecule. The bait molecule can be a potential drug if the investigator is trying to identify targets, or a target, such as an immobilized enzyme, if the investigator is screening for small molecules.[10] The bait is exposed to a mixture of potential binding partners, which can be identified after removing non-binding components.
Microbead-based immobilization
Microbead-based immobilization is a modular technique in that it allows the investigator to decide whether they wish to fish for protein targets from the proteome or drug-like compounds from chemical libraries. The macroscopic properties of microbeads make them amenable to relatively low labor enrichment applications, since they are easily to visualize and their bulk mass is readily removable protein solutions.[11] Microbeads were historically made of inert polymers, such as agarose and dextran, that are functionalized to attach a bait of choice. In the case of using proteins as bait, amine functional groups are common linkers to facilitate attachment. More modern approaches have benefitted from the popularization of dynabeads, a type of magnetic microbead, which enable magnetic separation of bead-immobilized analytes from treated samples. Magnetic beads exhibit superparamagnetic properties, which make them very easy to remove from solution using an external magnet.[12] In a simplified workflow, magnetic beads are used to immobilize a protein target, then the beads are mixed with a chemical library to screen for potential ligands. High-affinity ligands bind to the immobilized target and resist removal by washing, so they are enriched in the sample. Conversely, a ligand of interest can be immobilized and screened against proteome proteins by incubation with a lysate.[12]
Hybrid solution- and immobilization-based strategies have been applied, in which ligands functionalized with an enrichment tag, such as biotin, are allowed to float freely in solution and find their target proteins. After an incubation period, ligand-protein complexes can be reacted with streptavidin-coated beads, which bind the biotin tag and allow for pull-down and identification of interaction partners. This technology can be extended to assist with preparation of samples for ABPP and photoaffinity labeling. While immobilization approaches have been reproducible and successful, it is impossible to avoid the limitation of immobilization-induced steric hindrance, which interferes with induced fit. Another drawback is non-specific adsorption of both proteins and small molecules to the bead surface, which has the potential to generate false positives.[10]
Affinity chromatography
Affinity chromatography emerged in the 1950s as a rarely used method used to purify enzymes; it has since seen mainstream use and is the oldest among chemoproteomic approaches.[13] Affinity chromatography is performed following one of two basic formats: ligand immobilization or target immobilization. Under the ligand immobilization format, a ligand of interest - often a drug lead - is immobilized within a chromatography column and acts as the stationary phase. A complex sample consisting of many proteins, such as a cell lysate, is passed through the column and the target of interest binds to the immobilized ligand while other sample components pass through the column unretained. Under the target immobilization format, a target of interest - often a disease-relevant protein - is immobilized within a chromatography column and acts as the stationary phase. Pooled compound libraries are then passed through the column in an application buffer, ligands are retained through binding interactions with the stationary phase, and other compounds pass through the column unretained.[14] In both cases, retained analytes can be eluted from the column and identified using mass spectrometry. A table of elution strategies is provided below.
| Strategy | Description | Mechanism(s) | Advantages | Disadvantages |
|---|---|---|---|---|
| Non-specific elution | Bound ligands are released after a change in mobile phase composition shifts the local chemical environment of the stationary phase; protein conformation is changed, causing release of ligands; alternatively solvent polarity is decreased, causing bound ligands to preferentially distribute into the mobile phase. | pH; ionic strength; polarity. | Can be fine tuned
to elute specific proteins or ligands. |
Elution conditions may not be strong enough to elute unoptimized compounds. |
| Isocratic elution | The elution buffer is identical to the application buffer, ligands move readily through the column but at different speeds according to their underlying affinity for the target. | Transient interactions with stationary phase. | Allows for comparison of compound affinities. | Can be time consuming; requires continuous fraction collection. |
| Biospecific elution | Analytes are eluted by adding a high concentration of a high-affinity binding partner to the mobile phase, bound analytes are competed off of the stationary phase; the additive can be either a protein that scavenges bound ligands or a small molecule that displaces them. | Competitive binding. | Allows for concentration of analytes with immediate elution; high recovery. | Compounds not previously tested may have higher affinity than the elution additive. |
Derivatization-free approaches

While the approaches above have shown success, they are inherently limited by their need for derivatization, which jeopardizes the affinity of the interaction that derivatized compounds are said to emulate and introduces steric hindrance.[10] Immobilized ligands and targets are limited in their ability to move freely through space in a way that replicates the native protein-ligand interaction, and conformational change from induced fit is often limited when proteins or drugs are immobilized. Probe-based approaches also alter the three-dimensional nature of the ligand-protein interaction by introducing functional groups to the ligand, which can alter compound activity. Derivatization-free approaches aim to infer interactions by proxy, often through observations of changes to protein stability upon binding, and sometimes through chromatographic co-elution.[15]
The stability-based methods below are thought to work due to ligand-induced shifts in equilibrium concentrations of protein conformational states. A single protein type in solution may be represented by individual molecules in a variety of conformations, with many of them different from one another despite being identical in amino acid sequence. Upon binding a drug, the majority of ligand-bound protein enters an energetically favorable conformation, and moves away from the unpredictable distribution of less stable conformers.[16] Thus, ligand binding is said to stabilize proteins, making them resistant to thermal, enzymatic and chemical degradation. Some examples of stability-based derivatization-free approaches follow.
Thermal proteome profiling (TPP)
Thermal proteome profiling (also, Cellular Thermal Shift Assay) is recently popularized strategy to infer ligand-protein interactions from shifts in protein thermal stability induced by ligand binding. In a typical assay setup, protein-containing samples are exposed to a ligand of choice, then those samples are aliquoted and heated to separate individual temperature points. Upon binding to a ligand, a protein's thermal stability is expected to increase, so ligand-bound proteins will be more resistant to thermal denaturation. After heating, the amount of non-denatured protein remaining is analyzed using quantitative proteomics and stability curves are generated. Upon comparison to an untreated stability curve, the treated curve is expected to shift to the right, indicating that ligand-induced stabilization occurred. Historically, thermal proteome profiling has been assessed using a western blot against a known target of interest. With the advent of high resolution Orbitrap mass spectrometers, this type of experiment can be executed on a proteome-wide scale and stability curves can be generated for thousands of proteins at once. Thermal proteome profiling has been successfully performed in vitro, in situ, and in vivo. When coupled with mass spectrometry, this technique is referred to as the Mass Spectrometry Cellular Thermal Shift Assay (MS-CETSA).[17]
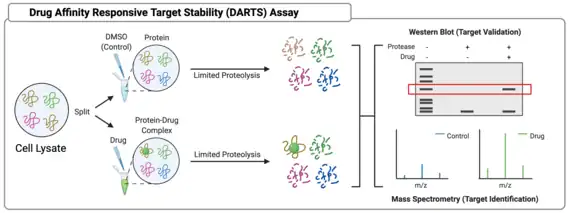
Drug affinity responsive target stability (DARTS)
The Drug Affinity Responsive Target Stability assay follows a similar basic assumption to TPP – that protein stability is increased by ligand binding. In DARTS, however, protein stability is assessed in response to digestion by a protease. Briefly, a sample of cell lysate is incubated with a small molecule of interest, the sample is split into aliquots, and each aliquot goes through limited proteolysis after addition of protease. Limited proteolysis is critical, since complete proteolysis would render even a ligand-bound protein completely digested. Samples are then analyzed via SDS-PAGE to assess differences in extent of digestion, and bands are then excised and analyzed via mass spectrometry to confirm the identities of proteins that are resist proteolysis. Alternatively, if the target is already suspected and is being tested for validation, a western blot protocol can be used to identify protein directly.[18]
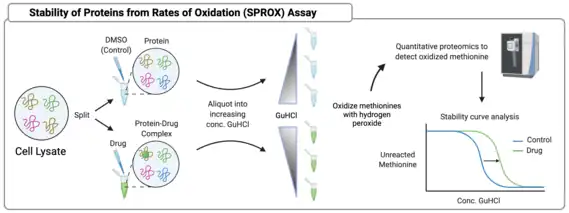
Stability of proteins from rates of oxidation (SPROX)
Stability of Proteins from Rates of Oxidation also rests upon the assumption that ligand binding confers protection to proteins from manners of degradation, this time from oxidation of methionine residues. In SPROX, a lysate is split and treated with drug or a DMSO control, then each group is further aliquoted into separate samples with increasing concentrations of the chaotrope and denaturant guanidinium hydrochloride (GuHCl). Depending on the concentration of GuHCl, proteins will unfold to varying degrees. Each sample is then reacted with hydrogen peroxide, which oxidizes methionine residues. Proteins that are stabilized by the drug will remain folded at higher concentrations of GuHCl and will experience less methionine oxidation. Oxidized methionine residues can be quantified via LC-MS/MS and used to generate methionine stability curves, which are a proxy for drug binding. There are drawbacks to the SPROX assay, namely that the only relevant peptides from SPROX samples are those with methionine residues, which account for approximately one-third of peptides, and for which there are currently no viable enrichment techniques. Only those methionines that are exposed to oxidation provide meaningful information, and not all differences in methionine oxidation are consistent with protein stabilization. Without enrichment, LC-MS/MS analysis of these peptides is challenging, as the contribution of other sample components to mass spectrometer noise can drown out relevant signal. Therefore, SPROX samples require fractionation to concentrate peptides of interest prior to LC-MS/MS analysis.[16]
Affinity selection-mass spectrometry
While adoption of affinity selection-mass spectrometry (AS-MS) has led to an expansion of assay formats,[19] the general technique follows a simple scheme. Protein targets are incubated with small molecules to allow for the formation of stable ligand-protein complexes, unbound small molecules are removed from the mixture, and the components of remaining ligand-protein complexes are analyzed using mass spectrometry.[19] The bound ligands identified are then categorized as hits and can be used to provide a starting point for lead generation. Since AS-MS measures binding in an unbiased manner, a hit does not need to be tied to a functional readout, opening the possibility of identifying drugs that act beyond active sites, such as allosteric modulators and chemical chaperones, all in a single assay.[20] Because small molecules can be directly identified by their exact mass, no derivatization is needed to confirm the validity of a hit.[20] Among derivatization- and label-free approaches, AS-MS has the unique advantage of being amenable to the assessment of multiple test compounds per experiment—as many as 20,000 compounds per experiment have been reported in the literature, and one group has reported assaying chemical libraries against heterogeneous protein pools.[20] The basic steps of AS-MS are described in more detail below.
.png.webp)
Affinity selection
A generalized AS-MS workflow begins with the pre-incubation of purified protein solutions (i.e. target proteins) with chemical libraries in microplates. Assays can be designed to contain sufficiently high protein concentrations to prevent competition for binding sites between structural analogs, ensuring that hits across a range of affinities can be identified; inversely, assays can contain low protein concentrations to allow for distinction between high and low affinity analogs and to inform structure-activity relationships.[20] The choice of a chemical library is less stringent than other high-throughput screening strategies owing to the lack of functional readouts, which would otherwise require deconvolution of the source compound that generates biological activity. Thus, the typical range for AS-MS is 400-3,000 compounds per pool.[20] Other considerations for screening are more practical, such as a need to balance desired compound concentration, which is usually in the micromolar range, with the fact that compound stock solutions are typically stored as 10 millimolar solutions, effectively capping the number of compounds screened in the thousands.[20] After appropriate test compounds and targets are selected and incubated, ligand-protein complexes can be separated by a variety of means.[19]
Separation of unbound small molecules and ligand-protein complexes
Affinity selection is followed by the removal of unbound small molecules via ultrafiltration or size-exclusion chromatography, making only protein-bound ligands available for downstream analysis.[19] Several types of ultrafiltration have been reported with varying degrees of throughput, including pressure-based, centrifugal, and precipitation-based ultrafiltration.[19] Under both pressure-based and centrifugal formats, unbound small molecules are forced through a semipermeable membrane that excludes proteins on the basis of size. Multiple washing steps are required after ultrafiltration to ensure complete removal of unbound small molecules.[19] Ultrafiltration can also be confounded by non-specific adsorption of unbound small molecules to the membrane.[19] A group at the University of Illinois published a screening strategy involving amyloid-beta, in which ligands were used to stabilize the protein and prevent its aggregation. Ultrafiltration was used to precipitate aggregated amyloid-beta and remove unbound ligands, while the ligand-stabilized protein was detected and quantified using mass spectrometry.[19]
Size-exclusion chromatography (SEC) is more widely used in industrial drug discovery and has the advantage of more efficient removal of unbound compounds as compared to ultrafiltration.[19] Size-exclusion approaches have been described in both high-performance liquid chromatography (HPLC) based and spin column formats.[19] In either case, a mixture of unbound ligands, proteins, ligand-protein complexes is passed through a column of porous beads. Ligand-protein complexes are excluded from entering the beads and exit the column quickly, while unbound ligands must travel through the beads and are retained by the column for a longer time. Ligands that elute from the column early on are therefore inferred to be bound to a protein.[19] The automated ligand identification system (ALIS), developed by Schering-Plough, uses a combined HPLC-based SEC to liquid chromatography-mass spectrometry (LC-MS) system that separates ligand-protein complexes from unbound ligands using SEC and diverts the complex toward an LC-MS system for on-line analysis of bound ligands.[19] Novartis' SpeedScreen uses SEC in 96-well spin column format, also known as gel filtration chromatography, which allows for simultaneous removal of unbound ligands from up to 96 samples.[21] Samples are also passed through porous beads, but centrifugation is used to move the sample through the column.[21] SpeedScreen is not coupled to an LC-MS system and requires further processing prior to final analysis. In this case, ligands must be freed from their targets and analyzed separately.
Analysis of bound ligands
The final step requires bioanalytical separation of bound ligands from their targets, and subsequent identification of ligands using liquid chromatography-mass spectrometry.[20] AS-MS offers means for identifying small molecule-protein interactions either directly - through top-down proteomic detection of intact complexes - or indirectly - through denaturation of small molecule-protein complexes followed by identification of small molecules using mass spectrometry.[19] The top-down approach requires direct infusion of the complex into an electrospray ionization mass spectrometry source under conditions gentle enough to preserve the interaction and maintain its integrity in the transition from liquid to gas.[19] While this was shown to be possible by Ganem and Henion in 1991, it is not suitable for high throughput.[19] Interestingly, electron capture dissociation, which is typically used in structure elucidation of peptides, has been used to identify ligand binding sites during top-down analysis.[19] A simpler method for analysis of bound ligands uses a protein precipitation extraction to denature proteins and release ligands into the precipitation solution, which can then be diluted and identified on an LC-MS system.
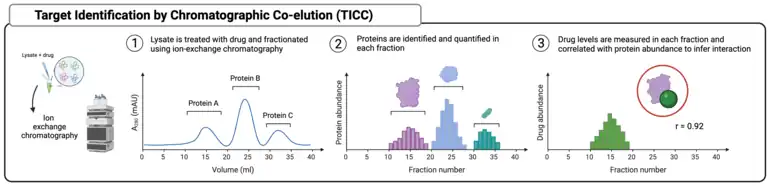
Target identification by chromatographic co-elution (TICC)
Target identification by chromatographic co-elution does not rely on differences in protein stability after drug treatment. Instead, it rests on the assumption that drugs form stable complexes with their target proteins, and that those complexes are robust enough to survive a chromatographic separation. In a typical workflow, a cell lysate is incubated with a drug, then the lysate is injected onto an ion-exchange chromatography system and fractionated. Lysate proteins are eluted along an ionic strength gradient and fractions are collected over short time intervals. Each fraction is analyzed by LC-MS/MS for both protein and drug content. Individual proteins elute with characteristic profiles, and pre-incubated drugs should mirror the elution profiles of the targets they complex with. Correlation of drug and protein elution profiles allows for targets to be narrowed down and inferred.[22]
Computational approaches
Molecular docking simulations
The development and application of bench-top chemoproteomics assays is often time consuming and cost-prohibitive. Molecular docking simulations have emerged as relatively low-cost, high-throughput means for ranking the strength of small molecule-protein interactions. Molecular docking requires accurate modeling of both ligand and protein conformation at atomic resolution, and is therefore aided by empirical determination of protein structure, often through orthogonal methods such as x-ray crystallography and cryogenic electron microscopy. Molecular docking strategies are categorized by the type of information that is already known about the ligand and protein of interest.[23]
Ligand-based methods
When a bioactive ligand with a known structure is to be screened against a protein with limited structural information, modeling is done with regard to ligand structure. Pharmacophore modeling identifies key electronic and structural features that are associated with therapeutic activity across similarly bioactive structural analogs, and accordingly requires large libraries with corresponding experimental data to enhance predictive power. Compound structures are superimposed virtually and common elements are scored on the basis of their tendency toward bioactivity.[24] The move away from lock-and-key based modeling toward induced-fit based modeling has improved binding predictions but has also given rise to the challenge of modeling ligand flexibility, which requires building a database of conformational models and utilizes large amounts of data storage space. Another approach is the so-called on-the-fly method, in which conformational models are tested during the process of pharmacophore modeling, without a database; this method requires significantly less storage space at the cost of high computing time.[24] A second challenge arises from the decision of how to superimpose analog structures. A common approach is to use a least-squares regression for superimposition, but this requires user-selected anchor points and therefore introduces human bias into the process. Pharmacophore models require training data sets, giving rise to another challenge—selection of the appropriate library of compounds to adequately train models. Data set size and chemical diversity significantly affect performance of the downstream product.[25]
Structure-based methods
Ideally, the structure of a drug target is known, which allows for structure-based pharmacophore modeling. A structure-based model integrates key structural properties of the protein's binding site, such as the spatial distribution of interaction points, with features identified from ligand based pharmacophore models to generate a holistic simulation of the ligand-protein interaction. A major challenge in structure-based modeling is to narrow down pharmacophore features, of which many are initially identified, to a set of high priority features, as modeling too many features is a computational challenge. Another challenge is the incompatibility of pharmacophore modeling with quantitative structure-activity relationship (QSAR) profiling. Accurate QSAR models rely on inclusion of many potential targets, not just the therapeutic target. For example, important pharmacophores may yield high-affinity interactions with therapeutic targets, but they may also lead to undesirable off-target activity, and they may also be substrates of metabolic enzymes, such as Cytochrome P450s. Therefore, pharmacophore modeling against therapeutic targets is only one component of the compound's total structure-activity relationship.[24]
Applications
Druggability
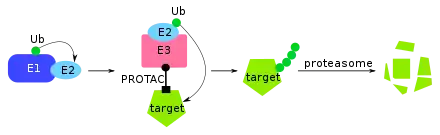
Chemoproteomic strategies have been used to expand the scope of druggable targets.[26] While historically successful drugs target well-defined binding pockets of druggable proteins, these define only about 15% of the annotated proteome.[26] To continue growing our pharmacopoeia, bold approaches to ligand discovery are required. The use of ABPP has coincidentally reinvigorated the search for newly ligandable sites. ABPP probes, intentionally used to label enzyme active sites, have been found to label many nucleophilic regions on many different proteins unintentionally. Originally thought to be experimental noise, these unintended reactions have clued researchers to the presence of sites that can potentially be targeted by novel covalent drugs. This is particularly salient in the case of proteins with no enzymatic activity to inhibit, or with mutated drug resistant proteins. In any of these cases, proteins can potentially be targeted for degradation using the novel drug modality of proteolysis-targeting-chimeras (PROTACs).[26] PROTACs are heterobifunctional small molecules that are designed to interact with a target and an E3 ubiquitin ligase. The interaction brings the E3 ubiquitin ligase close enough to the target that the target is labeled for degradation. The existence of potential covalent binding sites across the proteome suggests that many drugs can be covalently targeted using such a modality.[26]
Drug repurposing
Chemoproteomics is at the forefront of drug repurposing. This is particularly relevant in the era of COVID-19, which saw a dire need to rapidly identify FDA approved drugs that have antiviral activity.[27] In this context, a phenotypic screen is usually employed to identify drugs with a desired effect in vitro, such as inhibition of viral plaque formation. If a drug produces a positive test, the next step is to determine whether it is acting on a known or novel target. Chemoproteomics is thus a follow-up to phenotypic screening. In the case of COVID-19, Friman et al investigated off-target effects of the broad-spectrum antiviral Remdesivir, which was among the first repurposed drugs to be used in the pandemic.[27] Remdesivir was tested via thermal proteome profiling in a HepG2 cellular thermal shift assay, along with the controversial drug hydroxychloroquine, and investigators discovered TRIP13 as a potential off-target of Remdesivir.[27]
High-throughput screening
Approved drugs are never identified as hits in high-throughput screens because the chemical libraries used in screening have not been optimized against any targets. However, methods like affinity chromatography and affinity selection-mass spectrometry are workhorses of the pharmaceutical industry, and AS-MS particularly has been documented to produce a significant number of hits across many classes of difficult-to-drug proteins.[20] This is due in large part to the sheer volume of ligands that can be screened in a single assay. Researchers at the iHuman Institute at ShanghaiTech University employed of scheme in which 20,000 compounds per pool were screened against A2AR, a difficult G-protein coupled receptor to drug, with a 0.12% hit rate, leading to several high affinity ligands.[20]
| Technique | Bioactive Ligand | Target | Description | Reference |
|---|---|---|---|---|
| Activity based protein profiling | ABX1431 | MAGL | Cesar et al optimized MAGL inhibitor ABX1431 using a competitive inhibition system. | [28] |
| Photoaffinity labeling | Chloroquine | PfCRT | Lekostaj et al identified the chloroquine binding site on the Plasmodium chloroquine resistance transporter using photoaffinity labeling. | [7] |
| Affinity chromatography | Purvanolol | CDKs, CK1 | Knockaert et al confirmed CDKs and discovered CK1 as targets of purvanolol; CK1 is a Plasmodium protein, and purvanolol analogs have activity against Plasmodium. | [29] |
| Thermal proteome profiling (CETSA) | Remdesivir | TRIP13 | Friman et al identified TRIP13 as an off-target of Remdesivir after an investigation into off-target effects of repurposed COVID-19 drugs. | [27] |
| Drug affinity responsive target stability | Resveratrol | EIF4A | Lomenick et al discovered EIF4A as a target of the longevity-associated small molecule resveratrol. | [18] |
| Stability of proteins from rates of oxidation | Manassantin A | EEF1A1 | Geer Wallace et al discovered EEF1A1 as a target of the potent antineoplastic natural product Manassantin A | [15] |
| Affinity selection-mass spectrometry | Screening compounds | STING | Merck & Co. used AS-MS to successfully screen for ligands against the difficult to drug STING protein, which has a large binding site and strong competing endogenous ligand | [20] |
| Target identification by chromatographic co-elution | 4513-0042 | ERG6P | Chan et al identified the yeast protein ERG6P as a target of the antifungal natural product 4513-0042 and followed up with validation experiments | [22] |
References
- Wang, Lei (2020-08-21). "Chemical Tools and Mass Spectrometry-based Approaches for Exploring Reactivity and Selectivity of Small Molecules in Complex Proteomes". Doctoral Dissertations.
- Conway LP, Li W, Parker CG (March 2021). "Chemoproteomic-enabled phenotypic screening". Cell Chemical Biology. 28 (3): 371–393. doi:10.1016/j.chembiol.2021.01.012. PMID 33577749. S2CID 231910437.
- Moellering RE, Cravatt BF (January 2012). "How chemoproteomics can enable drug discovery and development". Chemistry & Biology. 19 (1): 11–22. doi:10.1016/j.chembiol.2012.01.001. PMC 3312051. PMID 22284350.
- Dowden H, Munro J (July 2019). "Trends in clinical success rates and therapeutic focus". Nature Reviews. Drug Discovery. 18 (7): 495–496. doi:10.1038/d41573-019-00074-z. PMID 31267067. S2CID 164564888.
- Yao, Xudong, ed. (2021). Advances in Chemical Proteomics (1st ed.). Elsevier. doi:10.1016/C2019-0-03583-2. ISBN 978-0-12-821433-6. S2CID 243825970.
- Nomura DK, Dix MM, Cravatt BF (September 2010). "Activity-based protein profiling for biochemical pathway discovery in cancer". Nature Reviews. Cancer. 10 (9): 630–638. doi:10.1038/nrc2901. PMC 3021511. PMID 20703252.
- Geoghegan KF, Johnson DS (2010). "Chemical Proteomic Technologies for Drug Target Identification". Annual Reports in Medicinal Chemistry. Elsevier. 45: 345–360. doi:10.1016/s0065-7743(10)45021-6. ISBN 9780123809025.
- Wang S, Tian Y, Wang M, Wang M, Sun GB, Sun XB (2018-04-09). "Advanced Activity-Based Protein Profiling Application Strategies for Drug Development". Frontiers in Pharmacology. 9: 353. doi:10.3389/fphar.2018.00353. PMC 5900428. PMID 29686618.
- Smith E, Collins I (February 2015). "Photoaffinity labeling in target- and binding-site identification". Future Medicinal Chemistry. 7 (2): 159–183. doi:10.4155/fmc.14.152. PMC 4413435. PMID 25686004.
- Chen X, Wang Y, Ma N, Tian J, Shao Y, Zhu B, et al. (May 2020). "Target identification of natural medicine with chemical proteomics approach: probe synthesis, target fishing and protein identification". Signal Transduction and Targeted Therapy. 5 (1): 72. doi:10.1038/s41392-020-0186-y. PMC 7239890. PMID 32435053.
- Hou X, Sun M, Bao T, Xie X, Wei F, Wang S (October 2020). "Recent advances in screening active components from natural products based on bioaffinity techniques". Acta Pharmaceutica Sinica B. 10 (10): 1800–1813. doi:10.1016/j.apsb.2020.04.016. PMC 7606101. PMID 33163336.
- Zhuo R, Liu H, Liu N, Wang Y (November 2016). "Ligand Fishing: A Remarkable Strategy for Discovering Bioactive Compounds from Complex Mixture of Natural Products". Molecules. 21 (11): 1516. doi:10.3390/molecules21111516. PMC 6274472. PMID 27845727.
- Turkov J (1978). Affinity Chromatography. Elsevier. ISBN 978-0-08-085812-8. OCLC 476215351.
- Rodriguez EL, Poddar S, Iftekhar S, Suh K, Woolfork AG, Ovbude S, et al. (November 2020). "Affinity chromatography: A review of trends and developments over the past 50 years". Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences. 1157: 122332. doi:10.1016/j.jchromb.2020.122332. PMC 7584770. PMID 32871378.
- Ha J, Park H, Park J, Park SB (March 2021). "Recent advances in identifying protein targets in drug discovery". Cell Chemical Biology. 28 (3): 394–423. doi:10.1016/j.chembiol.2020.12.001. PMID 33357463. S2CID 229693161.
- Lomenick B, Olsen RW, Huang J (January 2011). "Identification of direct protein targets of small molecules". ACS Chemical Biology. 6 (1): 34–46. doi:10.1021/cb100294v. PMC 3031183. PMID 21077692.
- Mateus A, Kurzawa N, Becher I, Sridharan S, Helm D, Stein F, et al. (March 2020). "Thermal proteome profiling for interrogating protein interactions". Molecular Systems Biology. 16 (3): e9232. doi:10.15252/msb.20199232. PMC 7057112. PMID 32133759.
- Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, et al. (December 2009). "Target identification using drug affinity responsive target stability (DARTS)". Proceedings of the National Academy of Sciences of the United States of America. 106 (51): 21984–21989. Bibcode:2009PNAS..10621984L. doi:10.1073/pnas.0910040106. PMC 2789755. PMID 19995983.
- Annis DA, Nickbarg E, Yang X, Ziebell MR, Whitehurst CE (October 2007). "Affinity selection-mass spectrometry screening techniques for small molecule drug discovery". Current Opinion in Chemical Biology. 11 (5): 518–526. doi:10.1016/j.cbpa.2007.07.011. PMID 17931956.
- Prudent R, Annis DA, Dandliker PJ, Ortholand JY, Roche D (2020-10-21). "Exploring new targets and chemical space with affinity selection-mass spectrometry". Nature Reviews Chemistry. 5 (1): 62–71. doi:10.1038/s41570-020-00229-2. PMID 37118102. S2CID 224811862.
- Zehender H, Le Goff F, Lehmann N, Filipuzzi I, Mayr LM (September 2004). "SpeedScreen: The "missing link" between genomics and lead discovery". Journal of Biomolecular Screening. 9 (6): 498–505. doi:10.1177/1087057104267605. PMID 15452336. S2CID 7135803.
- Chan JN, Vuckovic D, Sleno L, Olsen JB, Pogoutse O, Havugimana P, et al. (July 2012). "Target identification by chromatographic co-elution: monitoring of drug-protein interactions without immobilization or chemical derivatization". Molecular & Cellular Proteomics. 11 (7): M111.016642. doi:10.1074/mcp.M111.016642. PMC 3394955. PMID 22357554.
- Meng XY, Zhang HX, Mezei M, Cui M (June 2011). "Molecular docking: a powerful approach for structure-based drug discovery". Current Computer-Aided Drug Design. 7 (2): 146–157. doi:10.2174/157340911795677602. PMC 3151162. PMID 21534921.
- Yang SY (June 2010). "Pharmacophore modeling and applications in drug discovery: challenges and recent advances". Drug Discovery Today. 15 (11–12): 444–450. doi:10.1016/j.drudis.2010.03.013. PMID 20362693. S2CID 12843569.
- Khedkar SA, Malde AK, Coutinho EC, Srivastava S (March 2007). "Pharmacophore modeling in drug discovery and development: an overview". Medicinal Chemistry. 3 (2): 187–197. doi:10.2174/157340607780059521. PMID 17348856.
- Spradlin JN, Zhang E, Nomura DK (April 2021). "Reimagining Druggability Using Chemoproteomic Platforms". Accounts of Chemical Research. 54 (7): 1801–1813. doi:10.1021/acs.accounts.1c00065. PMID 33733731. S2CID 232303398.
- Haas P, Muralidharan M, Krogan NJ, Kaake RM, Hüttenhain R (February 2021). "Proteomic Approaches to Study SARS-CoV-2 Biology and COVID-19 Pathology". Journal of Proteome Research. 20 (2): 1133–1152. doi:10.1021/acs.jproteome.0c00764. PMC 7839417. PMID 33464917.
- Deng H, Lei Q, Wu Y, He Y, Li W (April 2020). "Activity-based protein profiling: Recent advances in medicinal chemistry". European Journal of Medicinal Chemistry. 191: 112151. doi:10.1016/j.ejmech.2020.112151. PMID 32109778. S2CID 211564093.
- Knockaert M, Gray N, Damiens E, Chang YT, Grellier P, Grant K, et al. (June 2000). "Intracellular targets of cyclin-dependent kinase inhibitors: identification by affinity chromatography using immobilised inhibitors". Chemistry & Biology. 7 (6): 411–422. doi:10.1016/s1074-5521(00)00124-1. PMID 10873834.
Branches of chemistry | |
|---|---|
| Analytical | |
| Theoretical | |
| Physical | |
| Inorganic | |
| Organic | |
| Biological | |
| Interdisciplinarity | |
| See also | |