Rhabdomyosarcoma
Rhabdomyosarcoma (RMS) is a highly aggressive form of cancer that develops from mesenchymal cells that have failed to fully differentiate into myocytes of skeletal muscle. Cells of the tumor are identified as rhabdomyoblasts.[1]
| Rhabdomyosarcoma | |
|---|---|
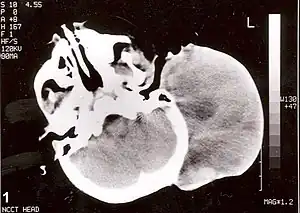 | |
| Non-contrast CT scan of head showing a large mass without any intracranial extension. The diagnosis was post-auricular congenital alveolar rhabdomyosarcoma. | |
| Specialty | Oncology |
The four subtypes are embryonal rhabdomyosarcoma, alveolar rhabdomyosarcoma, pleomorphic rhabdomyosarcoma, and spindle-cell/sclerosing rhabdomyosarcoma.[2] Embryonal and alveolar are the main groups, and these types are the most common soft tissue sarcomas of childhood and adolescence. The pleomorphic type is usually found in adults.[3]
It is generally considered to be a disease of childhood, as the vast majority of cases occur in those below the age of 18. It is commonly described as one of the small-blue-round-cell tumors of childhood due to its appearance on an H&E stain.[4] Despite being relatively rare, it accounts for approximately 40% of all recorded soft-tissue sarcomas.[5][6][7]
RMS can occur in any soft-tissue site in the body, but is primarily found in the head, neck, orbit, genitourinary tract, genitals, and extremities. No clear risk factors have been identified, but the disease has been associated with some congenital abnormalities.[5][8] Signs and symptoms vary according to tumor site, and prognosis is closely tied to the location of the primary tumor. Common sites of metastasis include the lungs, bone marrow, and bones.[9][10] There are many classification systems for RMS and a variety of defined histological types. Embryonal rhabdomyosarcoma is the most common type and comprises about 60% of cases.[11]
Outcomes vary considerably, with five-year survival rates between 35 and 95%, depending on the type of RMS involved, so clear diagnosis is critical for effective treatment and management.[11][12] Accurate and quick diagnosis is often difficult due to the heterogeneity of RMS tumors and a lack of strong genetic markers of the disease, although recent research by UVA Health researchers discovered "multiple lines of evidence supporting [the gene] AVIL is powerful driver for both major types of rhabdomyosarcoma," according to researcher Hui Li of the University of Virginia School of Medicine's Department of Pathology and UVA Cancer Center.[13] Malfunctions in AVIL, Li and his team found, play an essential role in the development of the two main subtypes of rhabdomyosarcoma. In a scientific paper outlining the findings, he and his colleagues describe rhabdomyosarcoma as "addicted" to the gene's excess activity. They ultimately label AVIL a "bona fide oncogene" for rhabdomyosarcoma.
Treatment usually involves a combination of surgery, chemotherapy, and radiation. 60 to 70% of newly diagnosed patients with nonmetastatic disease can be cured using this combined approach to therapy. Despite aggressive multimodality treatment, less than 20% of patients with metastatic RMS are able to be cured of their disease.[14]
Types
Given the difficulty in diagnosing rhabdomyosarcoma, definitive classification of subtypes has proven difficult. As a result, classification systems vary by institute and organization. Rhabdomyosarcoma in the 2020 WHO classification, though, is listed as four histological subtypes: embryonal, alveolar, pleomorphic, and spindle-cell/sclerosing.
Embryonal
Embryonal rhabdomyosarcoma (ERMS) is the most common histological variant, comprising about 60–70% of childhood cases. It is most common in children birth to four years old, with a maximum reported incidence of four cases per million children. ERMS is characterized by spindle-shaped cells with a stromal-rich appearance, and the morphology is similar to the developing muscle cells of a 6- to 8-week-old embryo. Tumors often present in the head and neck, as well as the genitourinary tract.
Embryonal subtype
Botryoid rhabdomyosarcoma is almost always found in mucosal-lined organs, including the vagina, bladder, and nasopharynx (although presentation in the nasopharynx typically affects older children). It often presents in infants younger than a year old, as a round, grape-like mass on the affected organ. Histologically, cells of the botryoid variant are defined by a dense tumor layer under an epithelium (cambium layer).[15] This subtype has a good prognosis.[11][12]
Botryoid rhabdomyosarcoma is also sometimes present in adult women, found in the cervix or uterus.[16]
Alveolar
Alveolar rhabdomyosarcoma (ARMS) is the second-most common type. ARMS comprises around 20–25% of RMS-related tumors, and it is equally distributed among all age groups with an incidence of about one case per million people ages 0 to 19. For this reason, it is the most common form of RMS observed in young adults and teenagers, who are less prone to the embryonal variant. This type of RMS is characterized by densely packed, round cells that arrange around spaces similar in shape to pulmonary alveoli, although variants have been discovered without these characteristic alveolar spacings. ARMS tends to form more often in the extremities, trunk, and peritoneum. It is also typically more aggressive than ERMS.[12][15]
Pleomorphic
Pleomorphic rhabdomyosarcoma (undifferentiated rhabdomyosarcoma), also known as anaplastic rhabdomyosarcoma, is defined by the presence of pleomorphic cells with large, lobate hyperchromatic nuclei and multipolar mitotic figures. These tumors display high heterogeneity and extremely poor differentiation. The pleomorphic cells may be diffuse or localized, with the diffuse variation correlating to a worse prognosis.[17] It occurs most often in adults, rarely in children, and is often discovered in the extremities.[10][18] Due to the lack of discernible separation among cancers of this type, clinicians often label undiagnosed sarcomas with little to no discernible features as anaplastic RMS. It is the most aggressive type of RMS, and often requires intensive treatment.[19]
Spindle-cell/sclerosing
Spindle-cell/sclerosing rhabdomyosarcoma is an added subtype listed in the 2020 WHO classification of soft-tissue sarcomas.[2]
This subtype is very similar to that of leiomyosarcoma (cancer of the smooth muscle tissue), and it has a fascicular, spindled, and leiomyomatous growth pattern with notable rhabdomyoblastic differentiation . It occurs most commonly in the paratesticular region, and the prognosis for this particular form of RMS is excellent with a reported five-year survival rate of 95%.[12] The sclerosing aspect of this subtype has a hyaline sclerosis and pseudovascular development.[20]
Multiple classification systems have been proposed for guiding management and treatment, and the most recent and widely used classification system is the "International Classification of Rhabdomyosarcoma" or ICR. It was created by the IRSG in 1995 after their series of four multi-institutional trials aimed at studying the presentation, histology, epidemiology, and treatment of RMS (IRSG I–IV).[11] The ICR system is based on prognostic indicators identified in IRSG I–IV. Pleomorphic rhabdomyosarcoma usually occurs in adults rather than children, and is therefore not included in this system.
Signs and symptoms
RMS can occur in almost any soft-tissue site in the body; the most common primary sites are genitourinary (24%), parameningeal (16%), extremity (19%), orbit (9%), other head and neck (10%), and miscellaneous other sites (22%).[15] RMS often presents as a mass, but signs and symptoms can vary widely depending on the site of the primary tumor. Genitourinary tumors may present with hematuria, urinary tract obstruction, and/or a scrotal or vaginal mass. Tumors that arise in the retroperitoneum and mediastinum can become quite large before producing signs and symptoms. Parameningeal tumors may present with cranial nerve dysfunction, symptoms of sinusitis, ear discharge, headaches, and facial pain. Orbital tumors often present with orbital swelling and proptosis. Extremity tumors generally present as a rapidly enlarging, firm mass in the relevant tissue. The cancer's prevalence in the head, face, and neck will often allow for earlier signs of the disease simply due to the obvious nature of tumors in these locations.[15] Despite the varying presentation and typically aggressive nature of the disease, RMS has the potential to be diagnosed and treated early. The fourth IRSG study found that 23% of patients were diagnosed in time for a complete resection of their cancer, and 15% had resection with only minimal remnants of the diseased cells.[21]
Risk factors
Rhabdomyosarcoma is difficult to diagnose. Risk factors that increase the likelihood of this cancer include inherited disorders such as Li-Fraumeni syndrome, Neurofibromatosis type 1, Beckwith-Wiedemann syndrome, Costello syndrome, Noonan syndrome,[22] and DICER1 syndrome.[23]
Genetic
There are multiple genetic lesions associated with rhabdomyosarcoma, but there has been little consistent data demonstrating an association between specific genetic abnormalities and outcome. However, alveolar and embryonal types of RMS can be distinguished cytogenetically, and identification of specific genetic lesions can allow for accurate classification of the ARMS subtype when the histopathological findings are equivocal or unclear. This is valuable for clinical practice as the alveolar type presents a higher risk to the patient and will often require more aggressive treatment than the embryonal type. Thus, ARMS is also referred to as Fusion Positive rhabdomyosarcoma (FP-RMS). Up to 90% of alveolar RMS cases present with a translocations of t(2;13)(q35, q14) or, less commonly, t(1;13)(p36, q15).[24][25] Both involve the translocation of a DNA binding domain of either PAX3[25] or PAX7[24], a member of the Paired Box family of transcription factors, to a transactivation site on FOXO1 (previously known as FKHR), a member of the forkhead/HNF-3 transcription factor family.[26] The t(2;13) translocation results in a fusion of the PAX3 gene with FOXO1, while the t(1;13) translocation involves the fusion of PAX7 with FOXO1.[27] PAX3 has a demonstrated role in muscle cell development, which supports its potential role in RMS. The t(2;13) translocation can result in the PAX3-FKHR fusion product, which is indicative of classic cystic ARMS.[27] Cases of FP-RMS are associated with a poorer prognosis than fusion-negative RMS.[28]
The fusion protein presents a potential therapeutic target, and in recent years more research has been conducted to clarify the role of PAX3-FOXO1 in FP-RMS. PAX3-FOXO1 is now known to drive key oncogenes such as MYC and MYCN by creating long-distance genetic interactions by super enhancers.[29] In this context, PAX3-FOXO1 both (1) drives the expression of MYC, MYCN and even MYOD1 (a transcription factor highly expressed in all RMS subtypes) but also (2) co-binds with these master transcription factors at super enhancers to support cancer growth.[29] Furthermore, it was demonstrated that FP-RMS subtypes were especially sensitive to inhibitors (such as JQ1) of a super enhancer bound protein BRD4.[29]
Embryonal RMS usually presents with a loss of heterozygosity (LOH) in the short arm of chromosome 11 (p11,15.5).[26][30] This region is associated with multiple oncogenes, and the potential loss-of-function of this region is likely associated with the loss of a tumor suppressor. However, the specific consequences of this LOH at (p11,15.5) have yet to be determined. The short arm of chromosome 11 is also the site of the insulin-like growth factor 2 gene (IGF-2), which is often over-expressed in RMS.
The loss-of-function of tumor suppressor p53 is associated with many cancers including rhabdomyosarcoma,[31] and approximately 50% of RMS cases have been shown to carry some form of mutation to the P53 gene. Other oncogenes often associated with rhabdomyosarcoma, albeit with less frequency, include NMYC, NRAS, KRAS, P16, and c-Met.[26][32] One study showed that 35% of embryonal RMS tumors contained activating mutations in either NRAS or KRAS and it is worth noting that ras activation has been shown to block myogenic differentiation, which could help explain its potential role in rhabdomyosarcogenesis.[33] More recently, a mechanistic and epigenetic link between mutant RAS isoforms and a block of myogenic differentiation has been demonstrated.[34] Furthermore, it has been shown that this differentiation block can be overcome with a clinical stage inhibitor of the MAP kinase pathway known as a MEK inhibitor.[34]
Diagnosis
Rhabdomyosarcoma is often difficult to diagnose due to its similarities to other cancers and varying levels of differentiation. It is loosely classified as one of the small-blue-round-cell tumors due to its appearance on an H&E stain. Other cancers that share this classification include neuroblastoma, Ewing sarcoma, and lymphoma, and a diagnosis of RMS requires confident elimination of these morphologically similar diseases.[15] The defining diagnostic trait for RMS is confirmation of malignant skeletal muscle differentiation with myogenesis (presenting as a plump, pink cytoplasm) under light microscopy.[5] Cross striations may or may not be present. Accurate diagnosis is usually accomplished through immunohistochemical staining for muscle-specific proteins such as myogenin, muscle-specific actin, desmin, D-myosin, and myoD1.[26][35][36] Myogenin, in particular, has been shown to be highly specific to RMS,[37] although the diagnostic significance of each protein marker may vary depending on the type and location of the malignant cells. The alveolar type of RMS tends to have stronger muscle-specific protein staining. Electron microscopy may also aid in diagnosis, with the presence of actin and myosin or Z bands pointing to a positive diagnosis of RMS.[5][35] Classification into types and subtypes is accomplished through further analysis of cellular morphology (alveolar spacings, presence of cambium layer, aneuploidy, etc.) as well as genetic sequencing of tumor cells. Some genetic markers, such as the PAX3-FKHR fusion gene expression in alveolar RMS, can aid in diagnosis. Open biopsy is usually required to obtain sufficient tissue for accurate diagnosis. All findings must be considered in context, as no one trait is a definitive indicator for RMS.
Staging
Following diagnosis and histopathological analysis, various imaging techniques may be used, including MRI, ultrasound, and a bone scan in order to determine the extent of local invasion and any metastasis. Further investigational techniques may be necessary depending on tumor sites. A parameningeal presentation of RMS will often require a lumbar puncture to rule out metastasis to the meninges. A paratesticular presentation will often require an abdominal CT to rule out local lymph node involvement, and so on. Outcomes are strongly tied to the extent of the disease, and its early mapping is important for treatment planning.
The current staging system for rhabdomyosarcoma is unusual relative to most cancers. It utilizes a modified TNM (tumor-nodes-metastasis) system originally developed by the IRSG.[11][12][38] This system accounts for tumor size (> or <5 cm), lymph node involvement, tumor site, and presence of metastasis.[15][38] It grades on a scale of 1 to 4 based on these criteria. In addition, patients are sorted by clinical group (from the clinical groups from the IRSG studies) based on the success of their first surgical resection.[38] The current Children's Oncology Group protocols for the treatment of RMS categorize patients into one of four risk categories based on tumor grade and clinical group, and these risk categories have been shown to be highly predictive of outcome.[35][39]
| Tumor site | Risk classification |
|---|---|
| Head and neck (orbit), biliary tract, genitourinary (excluding bladder and prostate) | Favorable |
| Cranial parameningial, bladder, extremities, prostate, other | Unfavorable |
Treatment
Treatment of rhabdomyosarcoma is a multidisciplinary practice involving the use of surgery, chemotherapy, radiation, and possibly immunotherapy. Surgery is generally the first step in a combined therapeutic approach. Resectability varies depending on tumor site, and RMS often presents in sites that don't allow for full surgical resection without significant morbidity and loss of function. Less than 20% of RMS tumors are fully resected with negative margins. Rhabdomyosarcomas are highly chemosensitive, with approximately 80% of cases responding to chemotherapy. In fact, multi-agent chemotherapy is indicated for all patients with rhabdomyosarcoma. Before the use of adjuvant and neoadjuvant therapy involving chemotherapeutic agents, treatment solely by surgical means had a survival rate of <20%. Modern survival rates with adjuvant therapy are approximately 60–70%.[8][40]
There are two main methods of chemotherapy treatment for RMS. There is the VAC regimen, consisting of vincristine, actinomycin D, and cyclophosphamide, and the IVA regimen, consisting of ifosfamide, vincristine, and actinomycin D. These drugs are administered in 9–15 cycles depending on the staging of the disease and other therapies used.[35] Other drug and therapy combinations may also show additional benefit. Addition of doxorubicin and cisplatin to the VAC regimen was shown to increase survival rates of patients with alveolar-type, early-stage RMS in IRS study III, and this same addition improved survival rates and doubled bladder salvage rates in patients with stage III RMS of the bladder.[17][35] In children and young adults with stage IV metastatic rhabdomyoscarcoma, a Cochrane review has found no evidence to support the use of high-dose chemotherapy as a standard therapy.[41]
Radiation therapy, which kill cancer cells with focused doses of radiation, is often indicated in the treatment of rhabdomyosarcoma, and the exclusion of this treatment from disease management has been shown to increase recurrence rates. Radiation therapy is used when resecting the entirety of the tumor would involve disfigurement or loss of important organs (eye, bladder, etc.). Generally, in any case where a lack of complete resection is suspected, radiation therapy is indicated.[15] Administration is usually following 6–12 weeks of chemotherapy if tumor cells are still present. The exception to this schedule is the presence of parameningeal tumors that have invaded the brain, spinal cord, or skull. In these cases radiation treatment is started immediately.[42][43] In some cases, special radiation treatment may be required. Brachytherapy, or the placement of small, radioactive "seeds" directly inside the tumor or cancer site, is often indicated in children with tumors of sensitive areas such as the testicles, bladder, or vagina. This reduces scattering and the degree of late toxicity following dosing.[44] Radiation therapy is more often indicated in higher stage classifications.
Immunotherapy is a more recent treatment modality that is still in development. This method involves recruiting and training the patient's immune system to target the cancer cells. This can be accomplished through administering small molecules designed to pull immune cells towards the tumors, taking immune cells pulled from the patient and training to attack tumors through presentation with tumor antigen, or other experimental methods. A specific example here would be presenting some of the patient's dendritic cells, which direct the immune system to foreign cells, with the PAX3-FKHR fusion protein in order to focus the patient's immune system to the malignant RMS cells. All cancers, including rhabdomyosarcoma, could potentially benefit from this new, immune-based approach.
Prognostic
Prognosis in rhabdomyosarcoma patients has been shown to be dependent on age, tumor site, resectability of tumor, tumor size, regional lymph node involvement, presence of metastasis, site and extent of metastasis, and biological and histopathological characteristics of the tumor cells.[45] Survival after recurrence is poor, and new salvage therapy strategies are needed.
Epidemiology
Rhabdomyosarcoma is the most common soft-tissue sarcoma in children as well as the third most common solid tumor in children. Recent estimates place the incidence of the disease at approximately 4.5 case per 1 million children/adolescents with approximately 250 new cases in the United States each year.[46][45] With the vast majority of cases of RMS occurring in children or adolescents, two-thirds of reported cases occur in youths under the age of 10.[5] RMS also occurs slightly more often in males than in females, with a ratio of approximately 1.3–1.5:1. In addition, slightly lower prevalence of the disease has been reported in black and Asian children relative to white children.[47][48][49] In most cases, there are no clear predisposing risk factors for the development of RMS. It tends to occur sporadically with no obvious cause. However, RMS has been correlated with familial cancer syndromes and congenital abnormalities including neurofibromatosis type 1,[50] Beckwith-Wiedemann syndrome,[51][52] Li–Fraumeni syndrome,[53] cardio-facio-cutaneous syndrome,[54] and Costello syndrome.[55] It has also been associated with parental use of cocaine and marijuana.[56]
History
Rhabdomyosarcoma was first described by Weber, a German physician, in 1845,[57] but it was not until the paper by Arthur Stout in 1946 that RMS was formally classified.[58] The first thirty years of investigation were conducted by the Intergroup Rhabdomyosarcoma Study Group (IRSG), an independent National Cancer Institute (NCI)-funded cooperative that has become a part of the Children's Oncology Group.
Research
Cancer stem cells of rhabdomyosarcoma have been identified and fibroblast growth factor receptor 3 has been suggested as their marker. Preclinical animal studies that try to use conditionally replicating adenoviruses against such cells are in progress.[59] Epigenetic therapy for rhabdomyosarcoma is becoming more important. A recent study by Bharathy et al. found that deacetylase inhibitor, entinostat works in aggressive subtype, alveolar rhabdomyosarcoma (aRMS) by specifically blocking the activity of HDAC3, thereby preventing epigenetic suppression of a microRNA that inhibits PAX3:FOXO1 translation. These findings and ongoing clinical trials (ADVL1513) shows promise for an effective therapy for some patients with aRMS.
See also
References
- "What Is Rhabdomyosarcoma?". www.cancer.org. Archived from the original on 2020-09-17. Retrieved 2021-02-11.
- Kallen, ME; Hornick, JL (January 2021). "The 2020 WHO Classification: What's New in Soft Tissue Tumor Pathology?". The American Journal of Surgical Pathology. 45 (1): e1–e23. doi:10.1097/PAS.0000000000001552. PMID 32796172. S2CID 225430576.
- Kumar, Vinay; Abbas, Abul K.; Aster, Jon C. (2018). Robbins basic pathology (Tenth ed.). Philadelphia, Pennsylvania. p. 830. ISBN 9780323353175.
{{cite book}}: CS1 maint: location missing publisher (link) - Chen QR, Vansant G, Oades K, et al. (February 2007). "Diagnosis of the small round blue cell tumors using multiplex polymerase chain reaction". The Journal of Molecular Diagnostics. 9 (1): 80–8. doi:10.2353/jmoldx.2007.060111. PMC 1867426. PMID 17251339. Archived from the original on 2020-02-13. Retrieved 2021-02-07.
- Arndt CA, Crist WM (July 1999). "Common musculoskeletal tumors of childhood and adolescence". N Engl J Med. 341 (5): 342–52. doi:10.1056/NEJM199907293410507. PMID 10423470.
- Maurer HM, Beltangady M, Gehan EA, et al. (January 1988). "The Intergroup Rhabdomyosarcoma Study-I. A final report". Cancer. 61 (2): 209–20. doi:10.1002/1097-0142(19880115)61:2<209::aid-cncr2820610202>3.0.co;2-l. PMID 3275486. S2CID 46247372.
- Maurer HM, Gehan EA, Beltangady M, et al. (March 1993). "The Intergroup Rhabdomyosarcoma Study-II". Cancer. 71 (5): 1904–22. doi:10.1002/1097-0142(19930301)71:5<1904::aid-cncr2820710530>3.0.co;2-x. PMID 8448756. S2CID 40778156.
- Pappo, A. S.; Shapiro, D. N.; Crist, W. M.; Maurer, H. M. (1995-08-01). "Biology and therapy of pediatric rhabdomyosarcoma". Journal of Clinical Oncology. 13 (8): 2123–2139. doi:10.1200/JCO.1995.13.8.2123. ISSN 0732-183X. PMID 7636557.
- Koscielniak E, Rodary C, Flamant F, et al. (1992). "Metastatic rhabdomyosarcoma and histologically similar tumors in childhood: a retrospective European multi-center analysis". Med Pediatr Oncol. 20 (3): 209–14. doi:10.1002/mpo.2950200305. PMID 1574030.
- Raney RB, Tefft M, Maurer HM, et al. (October 1988). "Disease patterns and survival rate in children with metastatic soft-tissue sarcoma. A report from the Intergroup Rhabdomyosarcoma Study (IRS)-I". Cancer. 62 (7): 1257–66. doi:10.1002/1097-0142(19881001)62:7<1257::aid-cncr2820620703>3.0.co;2-k. PMID 2843274. S2CID 22962204.
- Newton WA, Gehan EA, Webber BL, et al. (September 1995). "Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification--an Intergroup Rhabdomyosarcoma Study". Cancer. 76 (6): 1073–85. doi:10.1002/1097-0142(19950915)76:6<1073::aid-cncr2820760624>3.0.co;2-l. PMID 8625211. S2CID 23179823.
- Qualman, S. J.; Coffin, C. M.; Newton, W. A.; Hojo, H.; Triche, T. J.; Parham, D. M.; Crist, W. M. (1998-12-01). "Intergroup Rhabdomyosarcoma Study: update for pathologists". Pediatric and Developmental Pathology. 1 (6): 550–561. doi:10.1007/s100249900076. ISSN 1093-5266. PMID 9724344. S2CID 25785779.
- Xie, Zhongqiu; Janczyk, Pawel L.; Shi, Xinrui; Wang, Qiong; Singh, Sandeep; Cornelison, Robert; Xu, Jingjing; Mandell, James W.; Barr, Frederic G.; Li, Hui (2022-06-14). "Rhabdomyosarcomas are oncogene addicted to the activation of AVIL". Proceedings of the National Academy of Sciences. 119 (24): e2118048119. Bibcode:2022PNAS..11918048X. doi:10.1073/pnas.2118048119. ISSN 0027-8424. PMC 9214494. PMID 37146302.
- Hiniker, Susan M.; Donaldson, Sarah S. (2015-01-01). "Recent advances in understanding and managing rhabdomyosarcoma". F1000Prime Rep. 7: 59. doi:10.12703/P7-59. ISSN 2051-7599. PMC 4447051. PMID 26097732.
- Meyer, WH (2003). "Rhabdomyosarcoma". Holland-Frei Cancer Medicine (6th ed.). Hamilton (ON): BC Decker. Archived from the original on 2022-10-16. Retrieved 2017-09-10.
- Li, RF; Gupta, M; McCluggage, WG; Ronnett, BM (March 2013). "Embryonal rhabdomyosarcoma (botryoid type) of the uterine corpus and cervix in adult women: report of a case series and review of the literature". The American Journal of Surgical Pathology. 37 (3): 344–55. doi:10.1097/PAS.0b013e31826e0271. PMID 23348207. S2CID 26946298.
- Cecchetto, Giovanni; Bisogno, Gianni; De Corti, Federica; Dall'Igna, Patrizia; Inserra, Alessandro; Ferrari, Andrea; Garaventa, Alberto; Scagnellato, Angela; Carli, Modesto (2007-12-01). "Biopsy or debulking surgery as initial surgery for locally advanced rhabdomyosarcomas in children?: the experience of the Italian Cooperative Group studies". Cancer. 110 (11): 2561–2567. doi:10.1002/cncr.23079. ISSN 0008-543X. PMID 17941028. S2CID 25505511.
- Hays, D. M.; Lawrence, W.; Wharam, M.; Newton, W.; Ruymann, F. B.; Beltangady, M.; Maurer, H. M. (1989-01-01). "Primary reexcision for patients with 'microscopic residual' tumor following initial excision of sarcomas of trunk and extremity sites". Journal of Pediatric Surgery. 24 (1): 5–10. doi:10.1016/s0022-3468(89)80290-8. ISSN 0022-3468. PMID 2723995.
- Meza, Jane L.; Anderson, James; Pappo, Alberto S.; Meyer, William H.; Children's Oncology Group (2006-08-20). "Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: the Children's Oncology Group". Journal of Clinical Oncology. 24 (24): 3844–3851. doi:10.1200/JCO.2005.05.3801. ISSN 1527-7755. PMID 16921036.
- Folpe, Andrew L.; McKenney, Jesse K.; Bridge, Julia A.; Weiss, Sharon W. (2002-09-01). "Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma". The American Journal of Surgical Pathology. 26 (9): 1175–1183. doi:10.1097/00000478-200209000-00008. ISSN 0147-5185. PMID 12218574. S2CID 1792514.
- Crist, W. M.; Anderson, J. R.; Meza, J. L.; Fryer, C.; Raney, R. B.; Ruymann, F. B.; Breneman, J.; Qualman, S. J.; Wiener, E. (2001-06-15). "Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease". Journal of Clinical Oncology. 19 (12): 3091–3102. doi:10.1200/JCO.2001.19.12.3091. ISSN 0732-183X. PMID 11408506.
- "Risk Factors for Rhabdomyosarcoma". www.cancer.org. Archived from the original on 2022-08-06. Retrieved 2021-02-11.
- Robertson, JC; Jorcyk, CL; Oxford, JT (15 May 2018). "DICER1 Syndrome: DICER1 Mutations in Rare Cancers". Cancers. 10 (5): 143. doi:10.3390/cancers10050143. PMC 5977116. PMID 29762508.
- Davis RJ, D'Cruz CM, Lovell MA, Biegel JA, Barr FG (1994). "Fusion of PAX7 to FKHR by the Variant t(1;13)(p36;q14) Translocation in Alveolar Rhabdomyosarcoma". Cancer Research. 54 (11): 2869–2872. PMID 8187070. Archived from the original on 2016-08-10. Retrieved 2016-05-23.
- Barr, F. G.; Galili, N.; Holick, J.; Biegel, J. A.; Rovera, G.; Emanuel, B. S. (1993-02-01). "Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma". Nature Genetics. 3 (2): 113–117. doi:10.1038/ng0293-113. ISSN 1061-4036. PMID 8098985. S2CID 794511.
- Merlino, G.; Helman, L. J. (1999-09-20). "Rhabdomyosarcoma—working out the pathways". Oncogene. 18 (38): 5340–5348. doi:10.1038/sj.onc.1203038. ISSN 0950-9232. PMID 10498887. S2CID 23071344.
- Barr, F. G.; Chatten, J.; D'Cruz, C. M.; Wilson, A. E.; Nauta, L. E.; Nycum, L. M.; Biegel, J. A.; Womer, R. B. (1995-02-15). "Molecular assays for chromosomal translocations in the diagnosis of pediatric soft tissue sarcomas". JAMA. 273 (7): 553–557. doi:10.1001/jama.273.7.553. ISSN 0098-7484. PMID 7530783.
- Kelly, K. M.; Womer, R. B.; Sorensen, P. H.; Xiong, Q. B.; Barr, F. G. (1997-05-01). "Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma". Journal of Clinical Oncology. 15 (5): 1831–1836. doi:10.1200/JCO.1997.15.5.1831. ISSN 0732-183X. PMID 9164192.
- Gryder, Berkley E.; Yohe, Marielle E.; Chou, Hsien-Chao; Zhang, Xiaohu; Marques, Joana; Wachtel, Marco; Schaefer, Beat; Sen, Nirmalya; Song, Young (2017-08-01). "PAX3–FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability". Cancer Discovery. 7 (8): 884–899. doi:10.1158/2159-8290.CD-16-1297. ISSN 2159-8274. PMC 7802885. PMID 28446439.
- Visser, M.; Sijmons, C.; Bras, J.; Arceci, R. J.; Godfried, M.; Valentijn, L. J.; Voûte, P. A.; Baas, F. (1997-09-01). "Allelotype of pediatric rhabdomyosarcoma". Oncogene. 15 (11): 1309–1314. doi:10.1038/sj.onc.1201302. ISSN 0950-9232. PMID 9315099. S2CID 8160066.
- Oliner, J. D.; Kinzler, K. W.; Meltzer, P. S.; George, D. L.; Vogelstein, B. (1992-07-02). "Amplification of a gene encoding a p53-associated protein in human sarcomas" (PDF). Nature. 358 (6381): 80–83. Bibcode:1992Natur.358...80O. doi:10.1038/358080a0. hdl:2027.42/62637. PMID 1614537. S2CID 1056405. Archived from the original on 2022-10-16. Retrieved 2019-09-16.
- Weber-Hall, S.; Anderson, J.; McManus, A.; Abe, S.; Nojima, T.; Pinkerton, R.; Pritchard-Jones, K.; Shipley, J. (1996-07-15). "Gains, losses, and amplification of genomic material in rhabdomyosarcoma analyzed by comparative genomic hybridization". Cancer Research. 56 (14): 3220–3224. ISSN 0008-5472. PMID 8764111.
- Epstein, J. A.; Lam, P.; Jepeal, L.; Maas, R. L.; Shapiro, D. N. (1995-05-19). "Pax3 inhibits myogenic differentiation of cultured myoblast cells". The Journal of Biological Chemistry. 270 (20): 11719–11722. doi:10.1074/jbc.270.20.11719. ISSN 0021-9258. PMID 7744814. S2CID 25993698.
- Yohe, Marielle E.; Gryder, Berkley E.; Shern, Jack F.; Song, Young K.; Chou, Hsien-Chao; Sindiri, Sivasish; Mendoza, Arnulfo; Patidar, Rajesh; Zhang, Xiaohu (2018-07-04). "MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma". Science Translational Medicine. 10 (448): eaan4470. doi:10.1126/scitranslmed.aan4470. ISSN 1946-6234. PMC 8054766. PMID 29973406. S2CID 206694560.
- Dagher, R.; Helman, L. (1999-01-01). "Rhabdomyosarcoma: an overview". The Oncologist. 4 (1): 34–44. doi:10.1634/theoncologist.4-1-34. ISSN 1083-7159. PMID 10337369.
- Cessna, M. H.; Zhou, H.; Perkins, S. L.; Tripp, S. R.; Layfield, L.; Daines, C.; Coffin, C. M. (2001-09-01). "Are myogenin and myoD1 expression specific for rhabdomyosarcoma? A study of 150 cases, with emphasis on spindle cell mimics". The American Journal of Surgical Pathology. 25 (9): 1150–1157. doi:10.1097/00000478-200109000-00005. ISSN 0147-5185. PMID 11688574. S2CID 46691289.
- Kumar, S.; Perlman, E.; Harris, C. A.; Raffeld, M.; Tsokos, M. (2000-09-01). "Myogenin is a specific marker for rhabdomyosarcoma: an immunohistochemical study in paraffin-embedded tissues". Modern Pathology. 13 (9): 988–993. doi:10.1038/modpathol.3880179. ISSN 0893-3952. PMID 11007039. S2CID 21756898.
- Lawrence, W.; Anderson, J. R.; Gehan, E. A.; Maurer, H. (1997-09-15). "Pretreatment TNM staging of childhood rhabdomyosarcoma: a report of the Intergroup Rhabdomyosarcoma Study Group. Children's Cancer Study Group. Pediatric Oncology Group". Cancer. 80 (6): 1165–1170. doi:10.1002/(sici)1097-0142(19970915)80:6<1165::aid-cncr21>3.0.co;2-5. ISSN 0008-543X. PMID 9305719. S2CID 37393818.
- Pedrick, T. J.; Donaldson, S. S.; Cox, R. S. (1986-03-01). "Rhabdomyosarcoma: the Stanford experience using a TNM staging system". Journal of Clinical Oncology. 4 (3): 370–378. doi:10.1200/JCO.1986.4.3.370. ISSN 0732-183X. PMID 3950676.
- Ruymann, F. B.; Grovas, A. C. (2000-01-01). "Progress in the diagnosis and treatment of rhabdomyosarcoma and related soft tissue sarcomas". Cancer Investigation. 18 (3): 223–241. doi:10.3109/07357900009031827. ISSN 0735-7907. PMID 10754991. S2CID 25749451.
- Admiraal, Rick; van der Paardt, Marcel; Kobes, Jasmijn; Kremer, Leontien CM; Bisogno, Gianni; Merks, Johannes HM (2010-12-08). "High-dose chemotherapy for children and young adults with stage IV rhabdomyosarcoma". Cochrane Database of Systematic Reviews (12): CD006669. doi:10.1002/14651858.cd006669.pub2. ISSN 1465-1858. PMID 21154373.
- Raney, R. B.; Tefft, M.; Newton, W. A.; Ragab, A. H.; Lawrence, W.; Gehan, E. A.; Maurer, H. M. (1987-01-01). "Improved prognosis with intensive treatment of children with cranial soft tissue sarcomas arising in nonorbital parameningeal sites. A report from the Intergroup Rhabdomyosarcoma Study". Cancer. 59 (1): 147–155. doi:10.1002/1097-0142(19870101)59:1<147::aid-cncr2820590129>3.0.co;2-8. ISSN 0008-543X. PMID 3791141. S2CID 22713795.
- Raney, Richard Beverly; Meza, Jane; Anderson, James R.; Fryer, Christopher J.; Donaldson, Sarah S.; Breneman, John C.; Fitzgerald, Thomas J.; Gehan, Edmund A.; Michalski, Jeff M. (2002-01-01). "Treatment of children and adolescents with localized parameningeal sarcoma: experience of the Intergroup Rhabdomyosarcoma Study Group protocols IRS-II through –IV, 1978–1997". Medical and Pediatric Oncology. 38 (1): 22–32. doi:10.1002/mpo.1259. ISSN 0098-1532. PMID 11835233.
- Healey, E. A.; Shamberger, R. C.; Grier, H. E.; Loeffler, J. S.; Tarbell, N. J. (1995-05-15). "A 10-year experience of pediatric brachytherapy". International Journal of Radiation Oncology, Biology, Physics. 32 (2): 451–455. doi:10.1016/0360-3016(95)00520-9. ISSN 0360-3016. PMID 7772200.
- PDQ Pediatric Treatment Editorial Board (2002-01-01). Childhood Rhabdomyosarcoma Treatment (PDQ®): Health Professional Version. Bethesda (MD): National Cancer Institute (US). PMID 26389243. Archived from the original on 2022-08-19. Retrieved 2017-09-10.
- Ognjanovic, Simona; Linabery, Amy M.; Charbonneau, Bridget; Ross, Julie A. (2009-09-15). "Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005". Cancer. 115 (18): 4218–4226. doi:10.1002/cncr.24465. ISSN 0008-543X. PMC 2953716. PMID 19536876.
- Wexler, LH; Herman, LJ (1997). Principles and practice of pediatric oncology. 3rd ed. Philadelphia: Lippincott-Raven. pp. 799–829.
- Stiller, C. A.; McKinney, P. A.; Bunch, K. J.; Bailey, C. C.; Lewis, I. J. (1991-09-01). "Childhood cancer and ethnic group in Britain: a United Kingdom children's Cancer Study Group (UKCCSG) study". British Journal of Cancer. 64 (3): 543–548. doi:10.1038/bjc.1991.347. ISSN 0007-0920. PMC 1977662. PMID 1654982.
- Stiller, C. A.; Parkint, D. M. (1994-01-01). "International variations in the incidence of childhood soft-tissue sarcomas". Paediatric and Perinatal Epidemiology. 8 (1): 107–119. doi:10.1111/j.1365-3016.1994.tb00439.x. ISSN 1365-3016. PMID 8153013.
- Yang, P.; Grufferman, S.; Khoury, M. J.; Schwartz, A. G.; Kowalski, J.; Ruymann, F. B.; Maurer, H. M. (1995-01-01). "Association of childhood rhabdomyosarcoma with neurofibromatosis type I and birth defects". Genetic Epidemiology. 12 (5): 467–474. doi:10.1002/gepi.1370120504. ISSN 0741-0395. PMID 8557179. S2CID 23924644.
- Smith, A. C.; Squire, J. A.; Thorner, P.; Zielenska, M.; Shuman, C.; Grant, R.; Chitayat, D.; Nishikawa, J. L.; Weksberg, R. (2001-12-01). "Association of alveolar rhabdomyosarcoma with the Beckwith-Wiedemann syndrome". Pediatric and Developmental Pathology. 4 (6): 550–558. doi:10.1007/s10024001-0110-6. ISSN 1093-5266. PMID 11826361. S2CID 8824095.
- Cohen, P. R.; Kurzrock, R. (1995-01-01). "Miscellaneous genodermatoses: Beckwith-Wiedemann syndrome, Birt-Hogg-Dube syndrome, familial atypical multiple mole melanoma syndrome, hereditary tylosis, incontinentia pigmenti, and supernumerary nipples". Dermatologic Clinics. 13 (1): 211–229. doi:10.1016/S0733-8635(18)30121-9. ISSN 0733-8635. PMID 7712645. S2CID 36360747.
- Malkin, D.; Li, F. P.; Strong, L. C.; Fraumeni, J. F.; Nelson, C. E.; Kim, D. H.; Kassel, J.; Gryka, M. A.; Bischoff, F. Z. (1990-11-30). "Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms". Science. 250 (4985): 1233–1238. Bibcode:1990Sci...250.1233M. doi:10.1126/science.1978757. ISSN 0036-8075. PMID 1978757.
- Bisogno, G.; Murgia, A.; Mammi, I.; Strafella, M. S.; Carli, M. (1999-10-01). "Rhabdomyosarcoma in a patient with cardio-facio-cutaneous syndrome". Journal of Pediatric Hematology/Oncology. 21 (5): 424–427. doi:10.1097/00043426-199909000-00016. ISSN 1077-4114. PMID 10524458.
- Gripp, Karen W.; Scott, Charles I.; Nicholson, Linda; McDonald-McGinn, Donna M.; Ozeran, J. Daniel; Jones, Marilyn C.; Lin, Angela E.; Zackai, Elaine H. (2002-02-15). "Five additional Costello syndrome patients with rhabdomyosarcoma: proposal for a tumor screening protocol". American Journal of Medical Genetics. 108 (1): 80–87. doi:10.1002/ajmg.10241. ISSN 0148-7299. PMID 11857556.
- Grufferman, S.; Schwartz, A. G.; Ruymann, F. B.; Maurer, H. M. (1993-05-01). "Parents' use of cocaine and marijuana and increased risk of rhabdomyosarcoma in their children". Cancer Causes & Control. 4 (3): 217–224. doi:10.1007/bf00051316. ISSN 0957-5243. JSTOR 3552846. PMID 8318638. S2CID 26153806. Archived from the original on 2022-07-03. Retrieved 2020-03-29.
- Weber, CO (1854). "Anatomische Untersuchung einer hypertrophischen Zunge nebst Bemerkungen über die Neubildung quergestreifter Muskelfasern". Archiv für Pathologische Anatomie und Physiologie und für Klinische Medicin. 7: 115–125. doi:10.1007/BF01936232. S2CID 34545149. Archived from the original on 2022-11-14. Retrieved 2022-11-14.
- Stout, Arthur Purdy (1946-03-01). "Rhabdomyosarcoma of the Skeletal Muscles". Annals of Surgery. 123 (3): 447–472. doi:10.1097/00000658-194603000-00011. ISSN 0003-4932. PMC 1803493. PMID 17858752.
- Tanoue K (Jan 2014). "Survivin-responsive conditionally replicating adenovirus kills rhabdomyosarcoma stem cells more efficiently than their progeny". J Transl Med. 12: 27. doi:10.1186/1479-5876-12-27. PMC 3925355. PMID 24467821.
- Saboo, S. S.; Krajewski, K. M.; Zukotynski, K.; Howard, S.; Jagannathan, J. P.; Hornick, J. L.; Ramaiya, N. (2012). "Imaging Features of Primary and Secondary Adult Rhabdomyosarcoma". American Journal of Roentgenology. 199 (6): W694–W703. doi:10.2214/AJR.11.8213. PMID 23169742.