W. Clark Still
William Clark Still (born 1946) is an American organic chemist. As a distinguished professor at Columbia University, Clark Still made significant contributions to the field of organic chemistry, particularly in the areas of natural product synthesis, reaction development, conformational analysis, macrocyclic stereocontrol, and computational chemistry. Still and coworkers also developed the purification technique known as flash column chromatography which is widely used for the purification of organic compounds.[1]
W. Clark Still | |
|---|---|
| Born | 1946 Augusta, Georgia, United States |
| Nationality | American |
| Alma mater | Emory University |
| Known for | organic synthesis macrocyclic stereocontrol computational chemistry flash chromatography combinatorial chemistry |
| Awards | Alan T. Waterman Award (1981) Arthur C. Cope Scholar Award (1988) |
| Scientific career | |
| Fields | Organic chemistry |
| Institutions | Vanderbilt University Columbia University |
| Doctoral advisor | David Goldsmith |
| Doctoral students | Ge Li, Helma Wennemers |
Major contributions
Periplanone B
In 1979, Still reported the first total synthesis of periplanone B, the potent sex pheromone of the American cockroach.[2] Although the structural connectivity of this compound had been established spectroscopically, Still's synthesis confirmed the relative stereochemical relationships present in this natural product.[3] A key step in this synthesis makes use of an anionic oxy-Cope rearrangement to form the macrocyclic 10-membered ring found in periplanone B. In this transformation, the initial tertiary alcohol was treated with potassium hydride and 18-crown-6 at elevated temperature to induce the rearrangement, and the resulting enolate was trapped with chlorotrimethylsilane to form the macrocyclic silyl enol ether as a single diastereomer. This intermediate was then transformed into a key enone substrate over the course of five steps including Rubottom oxidation, TBS protection of the secondary alcohol, and formation of the exocyclic alkene via a selenoxide rearrangements.
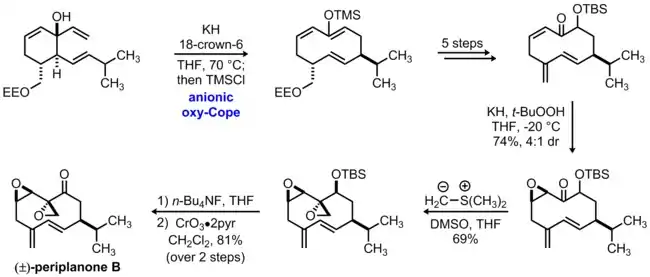
These transformations set the stage for the introduction of both epoxide groups, which were formed with high levels of diastereoselectivity based on the principles of macrocyclic stereocontrol. The first epoxide was introduced via nucleophilic epoxidation of the enone using potassium hydride and tert-butylhydroperoxide; this led to the formation of a 4:1 mixture of diastereomeric epoxides, favoring the desired isomer in which attack had occurred from the peripheral face of the enone. At this stage, the ketone was transformed into the second epoxide group using the Johnson-Corey-Chaykovsky reaction; notably, the product was formed as a single diastereomer, again due to initial peripheral attack of the dimethylsulfonium methylide nucleophile on the ketone. Subsequent removal of the TBS protecting group and Sarett oxidation of the resulting secondary alcohol completed the total synthesis of racemic periplanone B.
Monensin
In 1980, Still and coworkers reported the total synthesis of monensin, a complex polyether natural product that acts as an ionophore.[4][5] One of the key steps in this route is the late-stage union of two advanced fragments via an intermolecular aldol reaction. In this reaction, low-temperature deprotonation of the methyl ketone with lithium diisopropylamide formed the kinetic enolate which then underwent transmetalation upon the addition of magnesium bromide. Subsequent addition of the aldehyde component initiated a diastereoselective aldol reaction, forming the desired beta-hydroxyketone in 75% yield as a 3:1 mixture of diastereomers. Notably, this instance of acyclic stereocontrol is consistent with the Cram-Felkin-Anh model for the addition of nucleophiles to an aldehyde bearing an existing stereocenter in the alpha position.
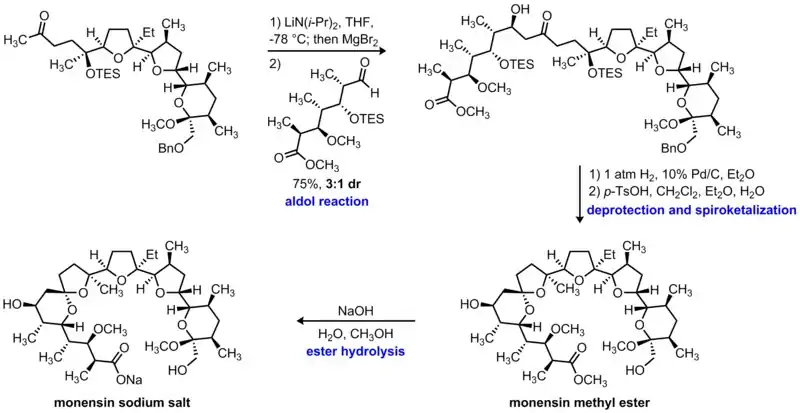
Having united these two complex fragments, Still was able to complete the synthesis of monensin in three additional steps. First, hydrogenolysis of the benzyl group afforded the free primary alcohol under standard conditions. Next, cleavage of the triethylsilyl (TES) protecting groups was achieved in the presence of para-toluenesulfonic acid; the resulting free secondary alcohols then engaged the ketone in a spontaneous cyclization reaction to form the thermodynamic spiroketal linkage. Finally, hydrolysis of the methyl ester under basic conditions furnished the sodium salt of monensin, completing the synthesis of this challenging natural product.
Wittig-Still rearrangement
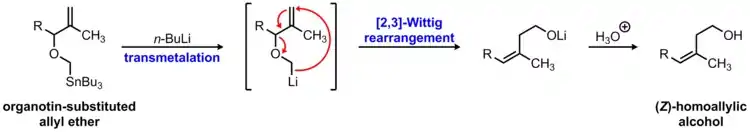
The 2,3-Wittig rearrangement is a useful carbon-carbon bond forming reaction that transforms an allyl ether into a homoallylic alcohol. However, a significant limitation of this reaction is the difficulty in forming the alkoxy-substituted carbanion which initiates the 2,3-sigmatropic rearrangement. Traditionally, this required the use of a strong base and the presence of a stabilizing group to enable deprotonation of the ether substrate. In 1978, Still dramatically increased the scope of the 2,3-Wittig rearrangement by introducing an organotin-substituted ether as a precursor to the alkoxy-substituted carbanion.[6] In this procedure, transmetalation of the organotin group can be achieved chemoselectively at low temperature using n-butyllithium to form an alpha-lithiated ether that undergoes the desired 2,3-sigmatropic rearrangement. This variation of the traditional 2,3-Wittig reaction is known as the Wittig-Still rearrangement.
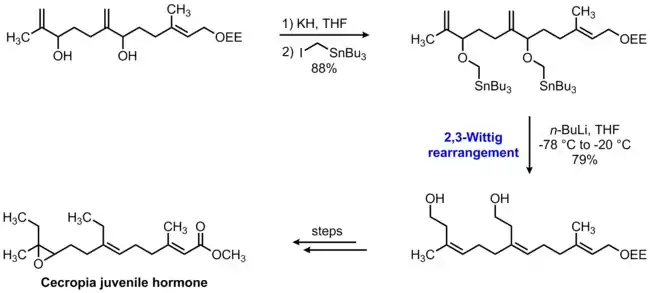
An application of the Wittig-Still rearrangement in the context of natural product synthesis can be seen in Still's 1979 formal synthesis of the Cecropia juvenile hormone.[7] The organotin-substituted ether that serves as the substrate for the rearrangement was readily prepared by bis-alkylation of the corresponding diol with iodomethyltributyltin. Treatment of this compound with two equivalents of n-butyllithium at low temperature resulted in transmetalation, initiating a double 2,3-sigmatropic rearrangement to give the bis-homoallylic alcohol product in 79% yield. Notably, this rearrangement is highly stereoselective with regard to the geometry of the trisubstituted olefins, giving exclusively the (Z,Z)-isomer shown.
Still-Gennari alkenation
The Horner-Wadsworth-Emmons reaction is a widely used olefination reaction in which a phosphonate-stabilized carbanion reacts with an aldehyde or ketone to form an alkene. In the standard HWE reaction, the phosphonate ester contains alkoxy substituents (typically methoxy or ethoxy), producing an E-alkene as the major product. In 1983, Still and Gennari reported that simply substituting the more electron-withdrawing 2,2,2-trifluoroethoxy groups on the phosphonate component reversed the stereoselectivity to produce predominantly Z-alkenes.[8] The use of a non-coordinating counterion was also found to be critical for high levels of Z-selectivity; this is typically accomplished by using a base with a potassium counterion in the presence of 18-crown-6. Taken together, this protocol is known as the Still-Gennari modification of the Horner-Wadsworth-Emmons reaction or, alternatively, as the Still-Gennari olefination.
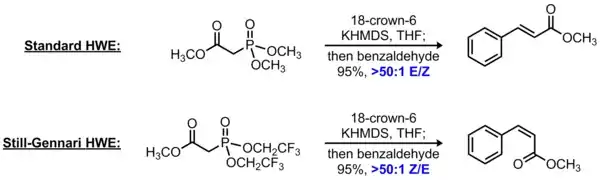
Although the mechanism of this reaction has not been fully elucidated, it is speculated that the use of an electron-deficient phosphonate accelerates elimination of the oxaphosphatane intermediate. This renders the initial addition of the phosphonate-stabilized carbanion to the aldehyde effectively irreversible, leading to Z-selectivity in analogy to the standard Wittig reaction.[9]
Flash column chromatography
In 1978, Still and coworkers published a highly influential paper reporting a purification technique known as flash column chromatography.[1] Prior to this report, column chromatography using silica gel as a stationary phase had already been established as a valuable method for the separation and purification of organic compounds. However, elution of the solvent by gravity alone was often a tedious process, requiring several hours and leading to poor separations due to band broadening via diffusion. Still's innovation was to apply pressure to the top of the column to increase the speed of solvent elution. Not only did this drastically reduce the time required to run the column, but it also allowed for the routine separation of compounds having an Rf difference of 0.10 or greater. After optimizing this procedure, Still compiled a table correlating column diameter, volume of eluant, amount of sample, and typical fraction size, providing a useful guide for application of this technique in the laboratory. Today, flash column chromatography is one of the most important methods for the purification of organic compounds, especially when working on a small scale (< 50 mg) where the techniques of recrystallization and distillation are impractical. Still's paper describing flash column chromatography remains his most highly cited publication and holds the distinction of being one of the most frequently downloaded papers from the Journal of Organic Chemistry, despite being published over 35 years ago.[10]
Computational chemistry and MacroModel
In addition to his contributions to synthetic organic chemistry, Clark Still was an early pioneer in applying computational methods to the study of organic compounds. Conformational analysis was integral to Still's study of macrocyclic stereocontrol, and there was a general need for a fast and reliable computational method for predicting an organic molecule's lowest energy conformation. In order to address the problem, Still and coworkers reported a simple Monte Carlo method for searching conformational space in 1989.[11] In this procedure, the internal coordinates for an organic molecule were varied at random, and the energy of the lowest energy conformation was determined after structure minimization. After each iteration, the minimum energy was compared to the values obtained previously in order to locate the global minimum; Still referred to this overall process as a Monte Carlo multiple-minimum search.
In an effort to make molecular modeling more accessible to practicing organic chemists, Still developed the integrated software program MacroModel in 1986 in association with Columbia University. Dr. Wayne Guida, a senior postdoctoral researcher in Still's group at the time, described the development of this software package in the following way:[12]
This was an extremely exciting time for us since we had funding from five pharmaceutical companies to actually produce a commercial product that would be used by their computational chemists and their synthetic chemists who might be interested in doing modeling as well. So, the software had to be accessible to the novice and expert alike and had to actually do something useful! Clark worked alongside the rest of us, and we all worked very hard to ensure that the molecular mechanics calculations churned out by our code were reasonably accurate and that the user interface was as intuitive as possible.
— Dr. Wayne Guida
One particularly notable feature of MacroModel was the inclusion of an implicit solvation model known as GB/SA (generalized Born model augmented with the hydrophobic solvent accessible surface area term).[13] The GB/SA model simulated solvent interactions with organic molecules by incorporating a continuous solvent field instead of including explicit individual solvent molecules in the computation. Still disclosed a full description of MacroModel in the Journal of Computation Chemistry in 1990,[14] and the rights to this software were later acquired by Schrodinger, Inc. in 1998.[15]
References
- Still, W. Clark; Kahn, M.; Mitra, A. (1978). "Rapid Chromatographic Technique for Preparative Separations With Moderate Separation". J. Org. Chem. 43 (14): 2923–2925. CiteSeerX 10.1.1.476.6501. doi:10.1021/jo00408a041.
- Still, W. Clark (1979). "Periplanone-B. Total synthesis and structure of the sex excitant pheromone of the American cockroach". J. Am. Chem. Soc. 101 (9): 2493–2495. doi:10.1021/ja00503a048.
- Nicolaou, K.C.; Sorensen, E.J. (1996). Classics in Total Synthesis. Weinheim: VCH. pp. 211–220.
- Still, W.C.; Collum, D.B; McDonald, J.H., III (1980). "Synthesis of the Polyether Antibiotic Monensin. 3. Coupling of Precursors and Transformation to Monensin". J. Am. Chem. Soc. 102 (6): 2120–2121. doi:10.1021/ja00526a075.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Nicolaou, K.C.; Sorensen, E.J. (1996). Classics in Total Synthesis. Weinheim: VCH. pp. 227–248.
- Still, W.C.; Mitra, A. (1978). "A highly stereoselective synthesis of Z-trisubstituted olefins via [2,3]-sigmatropic rearrangement. Preference for a pseudoaxially substituted transition state". J. Am. Chem. Soc. 100 (6): 1927–1928. doi:10.1021/ja00474a049.
- Still, W.C.; McDonald, J.H., III; Collum, D.; Mitra, A. (1979). "A Highly Stereoselective Synthesis of the C18 Cecropia Juvenile Hormone". Tetrahedron Lett. 20 (7): 593–594. doi:10.1016/S0040-4039(01)86010-X.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Still, W.C.; Gennari, C. (1983). "Direct synthesis of Z-unsaturated esters. A useful modification of the Horner-Emmons olefination". Tetrahedron Lett. 24 (41): 4405–4408. doi:10.1016/S0040-4039(00)85909-2.
- Kurti, Laszlo; Czako, Barbara (2005). Strategic Applications of Named Reactions in Organic Synthesis. Elsevier, Inc. pp. 214–215.
- "The Journal of Organic Chemistry: Most Read Articles". The American Chemical Society. Retrieved 26 May 2014.
- Still, W.C.; Chang, G.; Guida, W. (1989). "An internal-coordinate Monte Carlo method for searching conformational space". J. Am. Chem. Soc. 111 (12): 4379–4386. doi:10.1021/ja00194a035.
- "University of South Florida Chemistry News, Volume 5, No. 4, Autumn 2007" (PDF). Retrieved 27 May 2014.
- Still, W.C.; Tempczyk, A.; Hawley, R.; Hendrickson, T. (1990). "Semianalytical treatment of solvation for molecular mechanics and dynamics". J. Am. Chem. Soc. 112 (16): 6127–6129. doi:10.1021/ja00172a038. S2CID 29454695.
- Still, W.C.; Mohamadi, F.; Richards, N.; Guida, W.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T. (1990). "Macromodel—an integrated software system for modeling organic and bioorganic molecules using molecular mechanics". J. Comput. Chem. 11 (4): 440–467. doi:10.1002/jcc.540110405. S2CID 97538839.
- "MacroModel -- Versatile, Full-featured Program for Molecular Modeling". Schrodinger, Inc. Retrieved 27 May 2014.