Sensenbrenner syndrome
Sensenbrenner syndrome (OMIM #218330) is a rare (less than 20 cases reported by 2010) multisystem disease first described by Judith A. Sensenbrenner in 1975.[1] It is inherited in an autosomal recessive fashion, and a number of genes appear to be responsible. Three genes responsible have been identified: intraflagellar transport (IFT)122 (WDR10),[2] IFT43—a subunit of the IFT complex A machinery of primary cilia,[3] and WDR35 (IFT121: TULP4)[4]
| Sensenbrenner syndrome | |
|---|---|
| Other names | Cranioectodermal dysplasia |
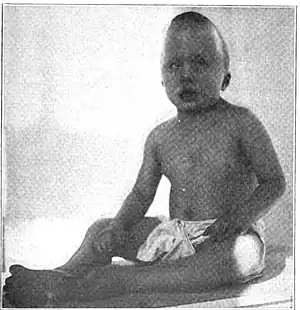 | |
| 6-year-old boy with symptoms of Sensenbrenner syndrome | |
It is also known as Sensenbrenner–Dorst–Owens syndrome, Levin syndrome I and cranioectodermal dysplasia (CED)
Presentation
These are pleomorphic and include
- dolichocephaly (with or without sagittal suture synostosis)
- microcephaly
- pre- and postnatal growth retardation
- brachydactyly
- narrow thorax
- rhizomelic dwarfism
- epicanthal folds
- hypodontia and/or microdontia
- sparse, slow-growing, hyperpigmented, fine hair
- nail dysplasia
- hypohydrosis
- chronic kidney failure
- heart defects
- liver fibrosis
- visual deficits
- photophobia
- hypoplasia of the posterior corpus callosum
- aberrant calcium homeostasis
Electroretinography shows gross abnormalities.
Two fetuses of 19 and 23 weeks gestation have also been reported.[5] They showed acromesomelic shortening, craniofacial characteristics with absence of craniosynostosis, small kidneys with tubular and glomerular microscopic cysts, persistent ductal plate with portal fibrosis in the liver, small adrenals, an enlarged cisterna magna and a posterior fossa cyst.
Cause

The gene IFT122 is located on the long arm of chromosome 3 (3q21-3q24). The gene lies on the Watson (plus) strand and is 80,047 bases in length. The encoded protein has 1241 amino acids and a predicted weight of 141.825 kiloDaltons (kDa). It is a member of the WD repeat protein family.
WDR35 is also a member of the WD repeat protein family. The gene is located on the short arm of chromosome 2 (2p24.1–2p24.3) The gene lies on the Crick (minus) strand and is 79,745 bases in length. The encoded protein is 1181 amino acids in length and its predicted molecular weight is 133.547 kiloDaltons.
The gene IFT43 lies on the Watson (plus) strand of the long arm of chromosome 14 (14q24.3).
A mouse model for IFT122 has been created.[6] Mutants deficient in IFT122 show multiple developmental defects (many are lethal), including exencephaly, situs viscerum inversus, delay in turning, hemorrhage and defects in limb development. In the node, primary cilia were absent or malformed in homozygous mutant and heterozygous embryos, respectively.
Impairment of the Sonic hedgehog pathway was apparent in both neural tube patterning (expansion of motoneurons and rostrocaudal level-dependent contraction or expansion of the dorsolateral interneurons) and limb patterning (ectrosyndactyly).
Pathophysiology
The IFT machinery is organized in two structural complexes — A and B. These complexes are involved in the coordinated movement of macromolecular cargo from the basal body along axonemal microtubules to the cilium tip and back again. The anterograde movement of IFT particles out to the distal tip of cilia and flagella is driven by kinesin-2 while the retrograde movement of particles back to the cell body is driven by cytoplasmic dynein 1b/2
The IFT-A protein complex is involved in retrograde ciliary transport. Disruption of IFT43 disturbs transport from the ciliary tip to the base. Anterograde transport in the opposite direction remains normal resulting in accumulation of the IFT complex B proteins in the ciliary tip.
Pathology
The visual defects are due to photoreceptor dystrophy. The chronic kidney failure is due to tubulointerstitial nephropathy. The liver fibrosis is secondary to ductal plate malformation.
Diagnosis
References
- Sensenbrenner JA, Dorst JP, Owens RP (1975). "New syndrome of skeletal, dental and hair anomalies". Birth Defects Orig. Artic. Ser. 11 (2): 372–9. PMID 1227553.
- Walczak-Sztulpa J, Eggenschwiler J, Osborn D, Brown DA, Emma F, Klingenberg C, Hennekam RC, Torre G, Garshasbi M, Tzschach A, Szczepanska M, Krawczynski M, Zachwieja J, Zwolinska D, Beales PL, Ropers HH, Latos-Bielenska A, Kuss AW (2010). "Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene". Am. J. Hum. Genet. 86 (6): 949–56. doi:10.1016/j.ajhg.2010.04.012. PMC 3032067. PMID 20493458.
- Arts HH, Bongers EM, Mans DA, van Beersum SE, Oud MM, Bolat E, Spruijt L, Cornelissen EA, Schuurs-Hoeijmakers JH, de Leeuw N, Cormier-Daire V, Brunner HG, Knoers NV, Roepman R (2011). "C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome". J. Med. Genet. 48 (6): 390–5. doi:10.1136/jmg.2011.088864. PMID 21378380. S2CID 6073572.
- Gilissen C, Arts HH, Hoischen A, Spruijt L, Mans DA, Arts P, van Lier B, Steehouwer M, van Reeuwijk J, Kant SG, Roepman R, Knoers NV, Veltman JA, Brunner HG (2010). "Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome". Am. J. Hum. Genet. 87 (3): 418–23. doi:10.1016/j.ajhg.2010.08.004. PMC 2933349. PMID 20817137.
- Konstantinidou AE, Fryssira H, Sifakis S, Karadimas C, Kaminopetros P, Agrogiannis G, Velonis S, Nikkels PG, Patsouris E (2009). "Cranioectodermal dysplasia: a probable ciliopathy". Am. J. Med. Genet. A. 149A (10): 2206–11. doi:10.1002/ajmg.a.33013. PMID 19760621. S2CID 31080779.
- Cortellino S, Wang C, Wang B, Bassi MR, Caretti E, Champeval D, Calmont A, Jarnik M, Burch J, Zaret KS, Larue L, Bellacosa A (2009). "Defective ciliogenesis, embryonic lethality and severe impairment of the Sonic Hedgehog pathway caused by inactivation of the mouse complex A intraflagellar transport gene Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4". Dev. Biol. 325 (1): 225–37. doi:10.1016/j.ydbio.2008.10.020. PMC 2645042. PMID 19000668.