Error threshold (evolution)
In evolutionary biology and population genetics, the error threshold (or critical mutation rate) is a limit on the number of base pairs a self-replicating molecule may have before mutation will destroy the information in subsequent generations of the molecule. The error threshold is crucial to understanding "Eigen's paradox".
The error threshold is a concept in the origins of life (abiogenesis), in particular of very early life, before the advent of DNA. It is postulated that the first self-replicating molecules might have been small ribozyme-like RNA molecules. These molecules consist of strings of base pairs or "digits", and their order is a code that directs how the molecule interacts with its environment. All replication is subject to mutation error. During the replication process, each digit has a certain probability of being replaced by some other digit, which changes the way the molecule interacts with its environment, and may increase or decrease its fitness, or ability to reproduce, in that environment.
Fitness landscape
It was noted by Manfred Eigen in his 1971 paper (Eigen 1971) that this mutation process places a limit on the number of digits a molecule may have. If a molecule exceeds this critical size, the effect of the mutations becomes overwhelming and a runaway mutation process will destroy the information in subsequent generations of the molecule. The error threshold is also controlled by the "fitness landscape" for the molecules. The fitness landscape is characterized by the two concepts of height (=fitness) and distance (=number of mutations). Similar molecules are "close" to each other, and molecules that are fitter than others and more likely to reproduce, are "higher" in the landscape.
If a particular sequence and its neighbors have a high fitness, they will form a quasispecies and will be able to support longer sequence lengths than a fit sequence with few fit neighbors, or a less fit neighborhood of sequences. Also, it was noted by Wilke (Wilke 2005) that the error threshold concept does not apply in portions of the landscape where there are lethal mutations, in which the induced mutation yields zero fitness and prohibits the molecule from reproducing.
Eigen's paradox
Eigen's paradox is one of the most intractable puzzles in the study of the origins of life. It is thought that the error threshold concept described above limits the size of self replicating molecules to perhaps a few hundred digits, yet almost all life on earth requires much longer molecules to encode their genetic information. This problem is handled in living cells by enzymes that repair mutations, allowing the encoding molecules to reach sizes on the order of millions of base pairs. These large molecules must, of course, encode the very enzymes that repair them, and herein lies Eigen's paradox, first put forth by Manfred Eigen in his 1971 paper (Eigen 1971).[1] Simply stated, Eigen's paradox amounts to the following:
- Without error correction enzymes, the maximum size of a replicating molecule is about 100 base pairs.
- For a replicating molecule to encode error correction enzymes, it must be substantially larger than 100 bases.
This is a chicken-or-egg kind of a paradox, with an even more difficult solution. Which came first, the large genome or the error correction enzymes? A number of solutions to this paradox have been proposed:
- Stochastic corrector model (Szathmáry & Maynard Smith, 1995). In this proposed solution, a number of primitive molecules of say, two different types, are associated with each other in some way, perhaps by a capsule or "cell wall". If their reproductive success is enhanced by having, say, equal numbers in each cell, and reproduction occurs by division in which each of various types of molecules are randomly distributed among the "children", the process of selection will promote such equal representation in the cells, even though one of the molecules may have a selective advantage over the other.
- Relaxed error threshold (Kun et al., 2005) - Studies of actual ribozymes indicate that the mutation rate can be substantially less than first expected - on the order of 0.001 per base pair per replication. This may allow sequence lengths of the order of 7-8 thousand base pairs, sufficient to incorporate rudimentary error correction enzymes.
A simple mathematical model
Consider a 3-digit molecule [A,B,C] where A, B, and C can take on the values 0 and 1. There are eight such sequences ([000], [001], [010], [011], [100], [101], [110], and [111]). Let's say that the [000] molecule is the most fit; upon each replication it produces an average of copies, where . This molecule is called the "master sequence". The other seven sequences are less fit; they each produce only 1 copy per replication. The replication of each of the three digits is done with a mutation rate of μ. In other words, at every replication of a digit of a sequence, there is a probability that it will be erroneous; 0 will be replaced by 1 or vice versa. Let's ignore double mutations and the death of molecules (the population will grow infinitely), and divide the eight molecules into three classes depending on their Hamming distance from the master sequence:



Hamming
distanceSequence(s) 0 [000] 1 [001]
[010]
[100]2 [110]
[101]
[011]3 [111]
Note that the number of sequences for distance d is just the binomial coefficient for L=3, and that each sequence can be visualized as the vertex of an L=3 dimensional cube, with each edge of the cube specifying a mutation path in which the change Hamming distance is either zero or ±1. It can be seen that, for example, one third of the mutations of the [001] molecules will produce [000] molecules, while the other two thirds will produce the class 2 molecules [011] and [101]. We can now write the expression for the child populations of class i in terms of the parent populations .




where the matrix 'w’ that incorporates natural selection and mutation, according to quasispecies model, is given by:

where is the probability that an entire molecule will be replicated successfully. The eigenvectors of the w matrix will yield the equilibrium population numbers for each class. For example, if the mutation rate μ is zero, we will have Q=1, and the equilibrium concentrations will be . The master sequence, being the fittest will be the only one to survive. If we have a replication fidelity of Q=0.95 and genetic advantage of a=1.05, then the equilibrium concentrations will be roughly . It can be seen that the master sequence is not as dominant; nevertheless, sequences with low Hamming distance are in majority. If we have a replication fidelity of Q approaching 0, then the equilibrium concentrations will be roughly . This is a population with equal number of each of 8 sequences. (If we had perfectly equal population of all sequences, we would have populations of [1,3,3,1]/8.)

![[n_{0},n_{1},n_{2},n_{3}]=[1,0,0,0]](../I/2b77707e33cb921bda49ec8653444ef681c57cf4.svg)
![[0.33,0.38,0.24,0.06]](../I/f1a0d8e9ac1cf04d4831f3f9f5ef4c5e15ad75bc.svg)
![[0.125,0.375,0.375,0.125]](../I/9d99733a62ed1458e1d1225b3f851bc52ee98496.svg)
If we now go to the case where the number of base pairs is large, say L=100, we obtain behavior that resembles a phase transition. The plot below on the left shows a series of equilibrium concentrations divided by the binomial coefficient . (This multiplication will show the population for an individual sequence at that distance, and will yield a flat line for an equal distribution.) The selective advantage of the master sequence is set at a=1.05. The horizontal axis is the Hamming distance d . The various curves are for various total mutation rates . It is seen that for low values of the total mutation rate, the population consists of a quasispecies gathered in the neighborhood of the master sequence. Above a total mutation rate of about 1-Q=0.05, the distribution quickly spreads out to populate all sequences equally. The plot below on the right shows the fractional population of the master sequence as a function of the total mutation rate. Again it is seen that below a critical mutation rate of about 1-Q=0.05, the master sequence contains most of the population, while above this rate, it contains only about of the total population.



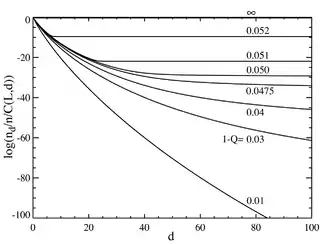
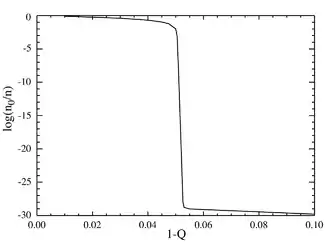
It can be seen that there is a sharp transition at a value of 1-Q just a bit larger than 0.05. For mutation rates above this value, the population of the master sequence drops to practically zero. Above this value, it dominates.
In the limit as L approaches infinity, the system does in fact have a phase transition at a critical value of Q: . One could think of the overall mutation rate (1-Q) as a sort of "temperature", which "melts" the fidelity of the molecular sequences above the critical "temperature" of . For faithful replication to occur, the information must be "frozen" into the genome.


References
- Holmes, Edward C. (2009). The Evolution and Emergence of RNA Viruses. Oxford University Press. pp. 22, 23, 48. ISBN 9780199211128. Retrieved 1 February 2019.
- Eigen, M. (1971). "Selforganization of matter and evolution of biological Macromolecules". Naturwissenschaften. 58 (10): 465–523. Bibcode:1971NW.....58..465E. doi:10.1007/BF00623322. PMID 4942363. S2CID 38296619.
- "Quasispecies theory in the context of population genetics - Claus O. Wilke" (PDF). Retrieved October 12, 2005.
- Campos, P. R. A.; Fontanari, J. F. (1999). "Finite-size scaling of the error threshold transition in finite populations" (PDF). J. Phys. A: Math. Gen. 32 (1): L1–L7. arXiv:cond-mat/9809209. Bibcode:1999JPhA...32L...1C. doi:10.1088/0305-4470/32/1/001. S2CID 16500591.
- Holmes, Edward C. (2005). "On being the right size". Nature Genetics. 37 (9): 923–924. doi:10.1038/ng0905-923. PMC 7097767. PMID 16132047.
- Eörs Szathmáry; John Maynard Smith (1995). "The major evolutionary transitions". Nature. 374 (6519): 227–232. Bibcode:1995Natur.374..227S. doi:10.1038/374227a0. PMID 7885442. S2CID 4315120.
- Luis Villarreal; Guenther Witzany (2013). "Rethinking quasispecies theory: From fittest type to cooperative consortia". World Journal of Biological Chemistry. 4 (4): 79–90. doi:10.4331/wjbc.v4.i4.79. PMC 3856310. PMID 24340131.
- Ádám Kun; Mauro Santos; Eörs Szathmáry (2005). "Real ribozymes suggest a relaxed error threshold". Nature Genetics. 37 (9): 1008–1011. doi:10.1038/ng1621. PMID 16127452. S2CID 30582475.