Degradomics
Degradomics is a sub-discipline of biology encompassing all the genomic and proteomic approaches devoted to the study of proteases, their inhibitors, and their substrates on a system-wide scale.[1][2] This includes the analysis of the protease and protease-substrate repertoires, also called "protease degradomes".[3] The scope of these degradomes can range from cell, tissue, and organism-wide scales.
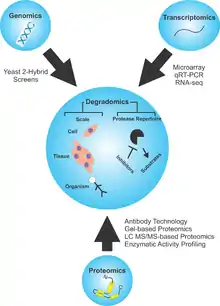
Background
As the second largest class of enzymes behind ubiquitin ligases and responsible for ~2% of any organism's genes,[4] proteases have drawn the attention of biologists to develop a field aimed to identify and quantify their roles in biology. First coined in 2000 by the Overall Lab in McQuibban et al.,[1] degradomics was described as linking proteases to substrates on a proteome basis. The discoveries of novel roles for proteases and breakthroughs in protease-substrate discovery would be summarized later by Dr. Carlos Lopez-Otin and Dr. Chris Overall,[2] introducing degradomics on a system-wide scale. They collated the current and emerging techniques available to describe proteolysis. By drawing attention to how proteolysis serves as an additional irreversible mechanism by which cells could achieve control over biological processes, they outlined the necessity of studying proteases for their functional relevance in processing bioactive molecules. These bioactive molecules play roles in coagulation, complement activation, DNA replication, cell-cycle control, cellular proliferation and migration, hemostasis, immunity, and apoptosis.[5][6] The degradome was broken down into two concepts, the first referring the entire profile of proteases expressed under by a cell, tissue, or organism under defined circumstances. The second definition applies specifically to the full substrate repertoire of a certain protease in a cell, tissue, or organism.
Dr. Overall's group would go on to annotate the complete human and mouse protease-inhibitor degradomes in 2003.[4] As the complexity of proteolytic networks was uncovered, more thorough descriptions of the degradome and ways to study it became necessary. In 2007, Dr. Overall updated the field with a review co-authored by Dr. Carl Blobel detailing how advanced methods were revolutionizing protease-substrate discovery.[3] They described the process of linking proteases to their substrates on a step by step process, beginning with biochemical and proteomic discovery, validation using cellular based assays, and progressing to whole organism levels using animal models. More recently, as technology and techniques have advanced, the Overall Lab and others have continued to direct the field using more powerful and quantitative techniques.[7][8][9]
Methods
Gene Microarrays
Traditionally DNA microarrays use complementary DNA or oligonucleotide probes to analyze messenger RNA (mRNA) from genes of interest. Extracted total RNA serves as a template for complementary DNA (cDNA) that is tagged with fluorescent probes before being allowed to hybridize to the microarray for visualization.[10] For proteases, specific probes for protease genes and their inhibitors have been developed to view expression patterns on the mRNA transcript level. The two platforms currently available for this purpose come from corporate and academic sources. Affymetrix's Hu/Mu ProtIn Microarray uses 516 and 456 probe sets to evaluate human and murine proteases, inhibitors, and interactors respectively.[11][12] CLIP-CHIP™, developed by the Overall Lab, is a complete protease and inhibitor DNA microarray for all 1561 human and murine proteases, non-proteolytic homologues, and their inhibitors.[13][14][15] Both of these tools allow comparison of expression patterns between normal and diseased samples and tissues. Unfortunately, as transcript levels often fail to reflect protein expression levels, gene microarrays are limited in representing protein in samples.[16] In addition, proteases recruited from remote sources like nearby tissues are ignored by these DNA based arrays, reiterating the need for protein based methods to confirm the presence and activity of functional enzymes when transcriptome analysis is performed.[2]
Quantitative real-time polymerase chain reaction (qRT-PCR)
A more sensitive approach to transcript analysis of a gene is quantitative real-time polymerase chain reaction (qRT-PCR), which has also seen action in quantifying protease mRNA levels. Again, total RNA is extracted and used to generate cDNA for PCR amplification. As long as there is a specific primer for a protease, protease inhibitor, or interacting molecule, qRT-PCR can serve as a highly sensitive method to detect minuscule amounts of mRNA copy numbers per cell.[17] A drawback that separates it from microarray analysis is its limited scope: a microarray can handle parallel analysis of multiple genes while qRT-PCR must amplify one mRNA for analysis at a time. It also suffers from the same limitation of microarray analysis regarding the lack of correlation between transcript and protein levels. However, its sensitivity lends it as a useful tool in validating microarray findings and quantifying specific protease transcripts of interest.
RNA sequencing (RNA-seq)
Whole transcriptome shotgun sequencing (WTSS)[18] is the latest in gene expression studies, using next generation sequencing (NGS) to quantify RNA in samples on a high throughput scale.[19] As biology trends toward using RNA-seq over microarray analysis in evaluating the transcriptome, so does degradomics. The field adapts the approach to analyzing the presence and quantity of transcripts of proteases, their substrates, and their inhibitors. While developed microarrays remain a major workhorse in studying gene expression in degradomics, its limitations of cross hybridization and dynamic range issues suggest RNA-seq will take a larger role as costs decrease and analysis improves.
Yeast Two-hybrid Screens
Yeast two-hybrid analyses have been adapted for protease-substrate discovery. As protease exosites play roles in protein-protein recognition and interaction, biologists have used exosites as tools to screen for protease interactors and potential substrates.[1] These protease exosite scanning assays use protease exosites as bait to scan a cDNA library for possible interacting partners.
Another early adaptation of yeast two-hybrid screening in protease-substrate discovery is Inactive-catalytic-domain capture (ICDC).[20] This approach attempts to avoid the limitation of protease exosite scanning, which fails to account for any substrates that do not require exosites to for recognition before cleavage. The bait for these assays are immobilized catalytically inactive mutant protease domains that cannot cleave and release their substrates once bound.
While useful in early degradomic studies, the limitations of adapted yeast two-hybrid screens have forced the field to move on to higher-throughput approaches for protease-substrate discovery. Their high rate of false positives and negatives,[21][22] inability to recognize complex interactions, lack of biologically compartmentalization, and failure to account for post-translational modifications necessary for protein-protein interactions hamper their usefulness.[23] Thus they have been largely replaced by proteomic methods as technology has improved.
Protease-specific Arrays
A protease specific protein array based on immobilized antibodies designed to capture specific proteases from biological samples offers a step up in analysis of protein levels beyond transcript expression. Capture antibodies spotted to nitrocellulose membranes can bind proteases in complex mixtures which have been pre-incubated and bound by detection antibodies allowing for parallel analysis of relative protease levels.[24] These arrays offer parallelization of protein levels over traditional western blot. Unfortunately, these assays fail to provide insight on enzymatic function for proteases and suffer similar drawbacks to western blots regarding reliable quantification.
Immunohistochemistry Approaches
As an antibody technique, immunohistochemistry (IHC) allows for validating protein presence.[23] It, and immunocytochemistry, allow for surveying the localization of proteases on a tissue or cellular scale respectively. It also can evaluate for the localization of cleavage products using monoclonal antibodies raised against neo-epitopes of cleavage sites produced by protease processing. Unfortunately, in addition to providing little functional information, IHC is also non-quantitative, making it an unappealing option for describing degradomics on system-wide scales.
Gel-based Proteomic Methods
Two-dimensional Polyacrylamide Electrophoresis (2D-PAGE) gels historically compared intensities from protease treated and untreated sample spots in order to identify possible candidate substrates.[25][26] A more recent improvement of this technique, fluorescent 2D difference gel electrophoresis (2D-DIGE), attempts to control standardization between gels for relative quantification.[27][28][29] Differentially labelling protease-treated and untreated samples with either Cy3 or Cy5, pooling said samples, and analyzing them together by 2D-PAGE allows substrate and cleavage products to be studied from the fluorescent gel. The spots corresponding to potentially substrate and cleavage products can be later elucidated using Mass Spectrometry or Edman Sequencing. The biggest drawbacks to using these techniques relate to the chemistry of the technique itself and its lack of sensitivity. As they rely on PAGE gels, extremely large, small, highly hydrophobic, acidic, or basic molecules will not be visualized.
Mass Spectrometry-based Proteomic Methods
Conventional shotgun proteomics identification of low abundance proteins in samples remains limited despite advances in Mass Spectrometry (MS) technology.[30] While abundant proteins can be easily detected, possible protease substrates of biological significance, such as cytokines, can be easily overlooked due to their low abundance. Most pre-clearing strategies designed to correct this also risk losing low abundant proteins, thus techniques designed specifically to target protease substrates for identification have been developed. These techniques have coalesced into a new field of positional proteomics or terminomics aimed at identifying protein N- or C-terminal modifications of protease substrates.[8] Terminomic approaches including Terminal Amine Isotopic Labeling of Substrates (TAILS) N-Terminomics, Combined FRActional Diagonal Chromatography (COFRADIC), and C-Terminomics add the level of stringency to conventional shotgun proteomics necessary to make them workhorse of degradomics.
TAILS, or “N-Terminomics,” was designed and developed by the Overall Lab to overcome the functional limitations of conventional proteomics by enriching both mature N-terminal peptides and newly generated N-terminal peptides of proteins produced by protease activity.[31][32] Formaldehyde or isobaric tags including Isotope-coded Affinity Tags (ICAT), 4 to 8 plex Isobaric tag for relative and absolute quantification (iTRAQ), or 10plex Tandem mass tags (TMT) block primary amines prior to trypsin digestion of proteome samples. The main step of the process is the negative selection of newly generated trypsin peptides using a specialized polymer. The polymer ignores the unreactive primary amines blocked by their tags, allowing them to be separated from trypsin generated peptides by ultrafiltration for Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) analysis.[31][33] These mature and neo-N-termini will differ in ratios between the protease treated versus untreated samples and make up the proteolytic fingerprint of a protease.[23] TAILS is also compatible with Stable isotope labeling by amino acids in cell culture (SILAC).
COFRADIC was the earliest technique to capitalize on negative selection to enrich for protein N-termini.[34] Sample proteins are first blocked by reduction and alkylation at their primary amines before endopeptidase treatment. Its negative selection method relies on strong cation exchange chromatography (SCX) to enrich for peptides representing N- and C-termini of proteins based on differences in peptide charge and pH.[35] Additional orthogonal chromatography treatments change the biochemical character of the peptides for further enrichment before final LC-MS/MS analysis. Groups have continued to adapt and improve this technology for protease-substrate discovery.[36]
C-terminomics has always been complicated due to the chemical nature of its targets.[8] Carboxyl groups are less reactive than primary amines, making C-terminomic techniques more complex than established N-terminomic approaches. However, adapted TAILS[37] and COFRADIC[38] workflows have been developed specifically to study the C-termini of proteins. Recently, the Overall Lab tackled another difficulty of C-terminomics, using endopeptidase LysargiNase™ to generate C-termini carrying N-terminal lysine or arginine residues.[39] Previously, C-termini lacked basic residues after endopeptidase digestion and could be missed in LC-MS/MS workflows.[8]
Another approach designed at further elucidating protease activity is Proteomic Identification of protease Cleavage Sites (PICS).[40] Beginning with a peptide library generated from endopeptidase digestion of a proteome, this technique allows for screening and characterizing the prime- and non-prime specificity for proteases. After digestion, primary amines and sulfhydryl are chemically blocked before digesting the sample again with the desired protease. Now, protease generated primary amines that constitute the prime site of cleavage can be biotinylated and isolated due to their reactivity and analyzed by LC-MS/MS. Non-prime sides sequences left behind must be determined using bioinformatics analysis of the extracted N-termini and full length protein sequences. These prime and non-prime sites give a full picture of protease cleavage site specificity.
Activity-based Profiling
To achieve functional degradomics, the enzymatic activity of proteases must be analyzed. Methods have been developed to distinguish the proteolytic activity of different enzymes in biological samples and separate active proteases from their inactive forms, namely zymogen precursors and those proteases bound by inhibitors.[2] Two techniques are activity-based probes (ABPs) and Proteolytic Signature Peptides (PSPs).
ABP molecules serve as probes to irreversibly bind only to active proteases and ignore their zymogen precursors[41][42] and inhibited proteases.[43] Placing a reactive group and a recognizable tag feature on the same molecule using a linker moiety gives an ABP molecule its structure.[44] The reactive molecule, designed after protease inhibitor mechanisms, lends ABPs their specificity towards targeting active proteases. Once bound, the reactive group acts much like an irreversible inhibitor to the protease.[23] Depending on the nature of the tag moiety, the ABP-protease complex can then be visualized or retrieved from biological samples for further studies of localization and quantification. Limitations including difficult production, specificity, stability, and toxicity[23] hamper ABP development but these probes have proved useful in revealing protease biological activity and remain a promising avenue in degradomic technology.[45][46][47]
PSPs do not depend on targeting active proteases with tagged compounds but rather on quantitative proteomics using stable isotope labeled standard peptides.[23] Standard peptides synthesized from amino acids labeled with stable isotope atoms serve as internal standards for serial dilutions of a sample. These allow for later absolute quantification of a proteins and post-translational modifications by mass spectrometry.[48] This technique has been adapted to absolute quantification of proteases, deciphering both activity states and total amounts in biological samples. This is thanks to trypsin treatment for mass spectrometry generating peptides specific to inactive zymogen precursors, active proteases, or common to both forms. PSPs is one form of standard peptide for absolute quantification and Standard of the Expressed Protease (STEP) is the other.[23] The major difference between the two is PSP sequences are designed to mimic tryptic peptides that contain sequences spanning the zymogen's pro-domain and the protease's final form, whereas STEP sequences match tryptic peptide sequences found in both forms of the protease. This capitalizes on protease activation, where the zymogen's pro-domain is cleaved off and the final form lacks that domain. Experiments using iTRAQ labeling and LC-MS/MS with STEP and PSP peptide internal standards have successfully quantified total and active protease levels in biological samples.[49] One major drawback for this approach is the inability to account for inhibitor bound enzymes. It is also difficult to ensure standard peptides can be generated for this method for each and every protease for study.[23]
Experiments using iTRAQ labeling and LC-MS/MS with STEP and PSP peptide internal standards have successfully quantified total and active protease levels in biological samples. One major drawback for this approach is the inability to account for inhibitor bound enzymes. It is also difficult to ensure standard peptides can be generated for this method for each and every protease for study. However, PSPs hold large potential for translating degradomics into clinical applications, as once a PSP is established it could aid in quantifying proteolytic signature biomarkers in Single Reaction Monitoring (SRM) and Multiple Reaction Monitoring (MRM) type clinical assays.
Bioinformatics
Owing to the increasing complexity of regulation of cellular processes and the roles proteases play in them, bioinformatics continues to be an invaluable tool for degradomics. Software, databases, and projects developed for this purpose have accompanied the advancement in technology. Software developed in the Overall Lab (CLIPPER) statistically evaluates cleavage site candidates determined by degradomic approaches.[50][51] One web-based data site, WebPICS,[52] incorporates and integrates cleavage site analysis from PICS experiments into MEROPS, the protease database. Another database, Termini oriented protein Function Inferred Database (TopFIND), serves as a knowledge base to integrate protein termini formed by protease processing with functional interpretations.[53] By combining research literature and other biological databases including UniProt, MEROPS, Ensembl, and TisDB, the database comprehensively renders protein termini modifications accessible to a broad scientific community. Using TopFIND, terminal modifications can be identified and visualized across proteins thanks to all available in silico, in vitro, and in vivo findings. Using TopFINDer and Path FINDer software, research findings can be mathematically modelled into a network of pathways regulated by proteases,[54] further contributing to the “protease web”.[55]
Significance and Impact
Protease Biology
Thanks to rapid advances in proteomics, genomics, and bioinformatics, protease research has been revolutionized. Degradomics emerged with the concept that proteolysis represents a specific mechanism for achieving cellular control over vital processes beyond control afforded by gene expression and translation and continues to produce the research necessary to understand the complex regulation of biology.[25] Where it was thought extracellular proteases degraded extracellular matrix (ECM), these proteases are now known to target and process a vast array of substrates with diverse roles, redefining protease functions and leading to a shift in interest towards new roles previously unknown to biology.[56] Degradomic studies of human tissue have also contributed to the Human Proteome Project (HPP) of the Human Proteome Organization (HUPO).[8]
Precision Medicine
The protease modulatory web represents opportunities to identify novel biomarkers for disease and targets for drug design.[55][56] Proteolytic processed N-termini have been proposed as potential biomarkers[57] as disease specific proteolysis has been well studied in pathologies such as inflammation [58] and cancer.[59] Contributions to degradomics have identified numerous characterized and novel protease substrates and continue to lead to speculation of previously unknown protease targets.[1][60] More recently, proteolytic signatures of cell death have been found using N-terminomic techniques on chemotherapy patient plasma samples.[61] Advancements in SRM and MRM clinical assays also allow for analyzing proteolytic signature biomarkers in patient samples and can be complemented by PSP quantification.[49] Deciphering these networks will aid drug design in understanding which substrates perform useful roles versus harmful ones to determine which should be targeted by drugs.[3]
References
- McQuibban, G.A., Gong J., Tam E.M., McCulloch C.A.G., Clark-Lewis I., Overall C.M.. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 289, 1202–1206 (2000).
- López-Otin, C., Overall, C.M. Protease degradomics: a new challenge for proteomics. Nat. Rev. Mol. Cell Biol. 3, 509–519 (2002).
- Overall C M., Blobel C. P. In search of partners: linking extracellular proteases to substrates. Nat. Rev. Mol. Cell Biol. 8, 245–257 (2007).
- Puente X. S., Sánchez L. M., Overall C. M., López-Otín C. Human and mouse proteases: a comparative genomic approach. Nature Reviews Genetics. 4(7):544–558 (2003).
- Barrett, A. J. et al. (eds) Handbook of Proteolytic Enzymes (Academic, London, 1998).
- Sternlicht, M. D., Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 17, 463–516 (2001).
- Marino G., Eckhard U., Overall C. M. Protein Termini and Their Modifications Revealed by Positional Proteomics. ACS Chem. Biol. 10, 1754-1764 (2015).
- Eckhard U., Marino G., Butler G. S., Overall C. M. Positional proteomics in the era of the human proteome project on the doorstep of precision medicine. Biochimie. 122, 110-118 (2016).
- Vizovisek M., Vidmar R., Fonovic M., Turk B. Current trends and challenges in proteomic identification of protease substrates. Biochimie. 122, 77-87 (2016).
- Yang YH, Speed T. Design issues for cDNA microarray experiments. Nat. Rev. Genet. 3, 579–588 (2002)
- Schwartz D.R., Moin K., Yao B., Matrisian L.M., Coussens L.M., Bugge T.H., Fingleton B., Acuff H.B., Sinnamon M., Nassar H., Platts A.E., Krawetz S.A., Linebaugh B.E., Sloane B.F. Hu/Mu ProtIn oligonucleotide microarray: dual-species array for profiling protease and protease inhibitor gene expression in tumors and their microenvironment. Mol. Cancer Res., 5 (5) 443–454 (2007).
- Acuff H.B., Sinnamon M., Fingleton B., Boone B., Levy S.E., Chen X., Pozzi A., Carbone D.P., Schwartz D.R., Moin K., Sloane B.F., Matrisian L.M. Analysis of host- and tumor-derived proteinases using a custom dual species microarray reveals a protective role for stromal matrix metalloproteinase-12 in non-small cell lung cancer Cancer Res., 66 (16) 7968–7975 (2006).
- Overall C. M., Tam E.M., Kappelhoff R., Connor A., Ewart T., Morrison C.J., Puente X., Lopez-Otin C., Seth A. Protease degradomics: mass spectrometry discovery of protease substrates and the CLIP-CHIP, a dedicated DNA microarray of all human proteases and inhibitors. Biol. Chem. 385, 493–504 (2004).
- Kappelhoff R., Auf dem Keller U., Overall C.M. Analysis of the degradome with the CLIP-CHIP microarray. Methods Mol. Biol.622:175-93 (2010).
- Kappelhoff R., Overall C.M. The CLIP-CHIP oligonucleotide microarray: dedicated array for analysis of all protease, nonproteolytic homolog, and inhibitor gene transcripts in human and mouse. Curr. Protoc. Protein Sci. 2007 Aug; Chapter 21:Unit 21.19.
- Lundberg E., Fagerberg L., Klevebring D., Matic I., Geiger T., Cox J., Algenas C., Lundeberg J., Mann M., Uhlen M. Defining the transcriptome and proteome in three functionally different human cell lines. Mol. Syst. Biol. 6:450 (2010).
- Nuttall R.K., Pennington C.J., Taplin J., Wheal A., Yong V.W., Forsyth P.A., Edwards D.R. Elevated membrane-type matrix metalloproteinases in gliomas revealed by profiling proteases and inhibitors in human cancer cells. Mol. Cancer Res. 1 (5) 333-345 (2003).
- Morin R., Bainbridge M., Fejes A., Hirst M., Krzywinski M., Pugh T., McDonald H., Varhol R., Jones S., Marra M. Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. Biotechniques 45 (1):81-94 (2008).
- Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10(1):57-63 (2009).
- Overall C. M., Tam E. M., Kappelhoff R., Connor A., Ewart T., Morrison C. J., Puente X., López-Otín C., and Seth A. Protease degradomics: mass spectrometry discovery of protease substrates and the CLIP-CHIP, a dedicated DNA microarray of all human proteases and inhibitors. Biol. Chem. 385, 493– 504 (2004).
- Uetz P., Giot L., Cagney G., Mansfield T. A., Judson R. S., Knight J. R., Lockshon D., Narayan V., Srinivasan M., Pochart P., Qureshi-Emili A., Li Y., Godwin B., Conover D., Kalbfleisch T., Vijayadamodar G., Yang M., Johnston M., Fields S., and Rothberg J. M. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403,623– 627 (2000).
- Ito T., Chiba T., Ozawa R., Yoshida M., Hattori M., Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. U. S. A. 98, 4569– 4574 (2001).
- Doucet A., Overall C.M. Protease proteomics: revealing protease in vivo functions using systems biology approaches Mol. Aspects Med., 29, 339–358 (2008).
- Kumar D., Moore R.M., Nash A., Springel E., Mercer B.M., Philipson E., Mansour J.M., Moore J.J. Decidual GM-CSF is a critical common intermediate necessary for thrombin and TNF induced in-vitro fetal membrane weakening. Placenta 35(12):1049-56 (2014).
- Hwang I.K., Park S.M., Kim S.Y., Lee S.T. A proteomic approach to identify substrates of matrix metalloproteinase-14 in human plasma. Biochim Biophys Acta. 1;1702(1):79-87 (2004).
- Lee A.Y., Goo Park S., Kho C.W., Young Park S., Cho S., Lee S.C., Lee D.H, Myung P.K., Park B.C. Identification of the degradome of Isp-1, a major intracellular serine protease of Bacillus subtilis, by two-dimensional gel electrophoresis and matrix- assisted laser desorption/ionization-time of flight analysis. Proteomics 4(11):3437-45 (2004).
- Zhou X.W., Blackman M.J., Howell S.A., Carruthers V.B. Proteomic analysis of cleavage events reveals a dynamic two-step mechanism for proteolysis of a key parasite adhesive complex. Mol. Cell Proteomics 3(6):565-76 (2004).
- Bredemeyer A.J., Lewis R.M., Malone J.P., Davis A.E., Gross J., Townsend R.R., Ley T.J. A proteomic approach for the discovery of protease substrates. Proc. Natl. Acad. Sci. U. S. A. 10;101(32):11785-90 (2004).
- Bech-Serra J.J., Santiago-Josefat B., Esselens C., Saftig P., Baselga J., Arribas J., Canals F. Proteomic identification of desmoglein 2 and activated leukocyte cell adhesion molecule as substrates of ADAM17 and ADAM10 by difference gel electrophoresis. Mol. Cell Biol. 26(13):5086-95 (2006).
- Anderson N.L., Anderson N.G. The human plasma proteome: history, character, and diagnostic prospects Mol. Cell. Proteom. MCP 1, 845–867(2002).
- Kleifeld O., Doucet A., auf dem Keller U., Prudova A., Schilling O., Kainthan R.K., et al. Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products Nat. Biotechnol. 28, 281–288 (2010).
- Prudova A., auf dem Keller U., Butler G.S., Overall C.M. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics Mol. Cell. Proteom. MCP 9, 894–911 (2010).
- Kleifeld O., Doucet A., Prudova A., auf dem Keller U., Gioia M., Kizhakkedathu J.N., et al. Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates Nat. Protoc., 6, 1578–1611 (2011).
- Gevaert K., Goethals M., Martens L., Van Damme J., Staes A., Thomas G.R., et al. Exploring proteomes and analyzing protein processing by mass spectrometric identification of sorted N-terminal peptides Nat. Biotechnol. 21, 566–569 (2003).
- Chen S.-H., Chen C.-R., Chen S.-H., Li D.-T., Hsu J.-L. Improved N(α)-acetylated peptide enrichment following dimethyl labeling and SCX J. Proteom. Res. 12, 3277–3287 (2013).
- Venne A.S., Solari F.A., Faden F., Paretti T., Dissmeyer N., Zahedi R.P. An improved workflow for quantitative N-terminal charge-based fractional diagonal chromatography (ChaFRADIC) to study proteolytic events in Arabidopsis thaliana. Proteomics (2015).
- Schilling O., Barré O., Huesgen P.F., Overall C.M. Proteome-wide analysis of protein carboxy termini: C terminomics Nat. Methods, 7, 508–511 (2010)
- Van Damme P., Staes A., Bronsoms S., Helsens K., Colaert N., Timmerman E., et al. Complementary positional proteomics for screening substrates of endo- and exoproteases Nat. Methods, 7, 512–515 (2010).
- Huesgen P.F., Overall C.M. N- and C-terminal degradomics: new approaches to reveal biological roles for plant proteases from substrate identification Physiol. Plant, 145, 5–17 (2012).
- Schilling O., Overall C.M. Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat Biotechnology 26(6) 685- 94 (2008).
- Williams E.B., Krishnaswamy S., Mann K.G. Zymogen/enzyme discrimination using peptide chloromethyl ketones J. Biol. Chem., 264 (13) 7536–7545 (1989).
- Kidd D., Liu Y., Cravatt B.F. Profiling serine hydrolase activities in complex proteomes Biochemistry, 40 (13) 4005–4015 (2001).
- Liu Y., Patricelli M.P., Cravatt B.F. Activity-based protein profiling: the serine hydrolases Proc. Natl. Acad. Sci. U.S.A., 96 (26) 14694–14699 (1999).
- Schmidinger H., Hermetter A., Birner-Gruenberger R. Activity-based proteomics: enzymatic activity profiling in complex proteomes Amino Acids, 30 (4) 333–350 (2006).
- Greenbaum D.C., Baruch A., Grainger M., Bozdech Z., Medzihradszky K.F., Engel J., DeRisi J., Holder A.A., Bogyo M. A role for the protease falcipain 1 in host cell invasion by the human malaria parasite Science, 298, (5600) 2002–2006 (2002).
- Winssinger N., Ficarro S., Schultz P.G., Harris J.L. Profiling protein function with small molecule microarrays Proc. Natl. Acad. Sci. U.S.A., 99 (17) 11139–11144 (2002).
- Saghatelian A., Jessani N., Joseph A., Humphrey M., Cravatt B.F. Activity-based probes for the proteomic profiling of metalloproteases Proc. Natl. Acad. Sci. U.S.A., 101 (27) 10000–10005 (2004).
- Gerber S.A., Rush J., Stemman O., Kirschner M.W., Gygi S.P. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS Proc. Natl. Acad. Sci. U.S.A., 100 (12) 6940–6945 (2003).
- Fahlman R.P., Chen W., Overall C.M. Absolute proteomic quantification of the activity state of proteases and proteolytic cleavages using proteolytic signature peptides and isobaric tags. J. Proteomics. 4;100:79-91 (2014).
- auf dem Keller U., Prudova A., Gioia M., Butler G.S., Overall C.M. A statistics-based platform for quantitative N-terminome analysis and identification of protease cleavage products. Mol. Cell. Proteomics 9, 912 – 927 (2010).
- auf dem Keller U., Overall C.M. CLIPPER: an add-on to the Trans-Proteomic Pipeline for the automated analysis of TAILS N-terminomics data. Biol Chem. 393(12):1477-83 (2012).
- Schilling O., auf dem Keller U., Overall C.M. Protease Specificity Profiling by Tandem Mass Spectrometry Using Proteome-derived Peptide Libraries. Gel-Free Proteomics: Methods and Protocols 753 257-272 (2011).
- Lange P.F., Huesgen P.F., Overall C.M. TopFIND 2.0 — Linking protein termini with proteolytic processing and modifications altering protein function. Nucleic Acids Res. 40, D351 – 361 (2012).
- Fortelny N., Yang S., Pavlidis P., Lange P.F., Overall C.M.. Proteome TopFIND 3.0 with TopFINDer and PathFINDer: database and analysis tools for the association of protein termini to pre- and post-translational events. Nucleic Acids Res. 43(Database issue):D290-7 (2015).
- Doucet A., Overall C.M. Protease proteomics: revealing protease in vivo functions using systems biology approaches. Mol. Aspects Med. 29(5):339-58 (2008).
- Butler G.S., Overall C.M. Proteomic validation of protease drug targets: pharmacoproteomics of matrix metalloproteinase inhibitor drugs using isotope-coded affinity tag labelling and tandem mass spectrometry. Curr. Pharm. Des.13(3):263-70 (2007).
- Huesgen P.F., Lange P.F., Overall C.M. Ensembles of protein termini and specific proteolytic signatures as candidate biomarkers of disease. Proteom. Clin. Appl., 8 338–350 (2014).
- auf dem Keller U., Prudova A., Eckhard U., Fingleton B., Overall C.M. Systems-level analysis of proteolytic events in increased vascular permeability and complement activation in skin inflammation. Sci. Signal, 6 p. rs2 (2013).
- Doucet A., Butler G.S., Rodríguez D., Prudova A., Overall C.M. Metadegradomics: toward in vivo quantitative degradomics of proteolytic post-translational modifications of the cancer proteome. Mol. Cell. Proteom. MCP, 7 1925–1951 (2008).
- Starr A.E., Dufour A., Maier J., Overall C.M. Biochemical analysis of matrix metalloproteinase activation of chemokines CCL15 and CCL23 and increased glycosaminoglycan binding of CCL16. J. Biol. Chem., 287 5848–5860 (2012).
- Wiita A.P., Hsu G.W., Lu C.M., Esensten J.H., Wells J.A.. Circulating proteolytic signatures of chemotherapy-induced cell death in humans discovered by N-terminal labeling. Proc. Natl. Acad. Sci. U. S. A., 111 7594–7599 (2014).