Diastematomyelia
Diastematomyelia (occasionally diastomyelia) is a congenital disorder in which a part of the spinal cord is split, usually at the level of the upper lumbar vertebra in the longitudinal (sagittal) direction. Females are affected much more commonly than males. This condition occurs in the presence of an osseous, cartilaginous or fibrous septum in the central portion of the spinal canal which then produces a complete or incomplete sagittal division of the spinal cord into two hemicords. When the split does not reunite distally to the spur, the condition is referred to as diplomyelia, which is true duplication of the spinal cord.[1]
| Diastematomyelia | |
|---|---|
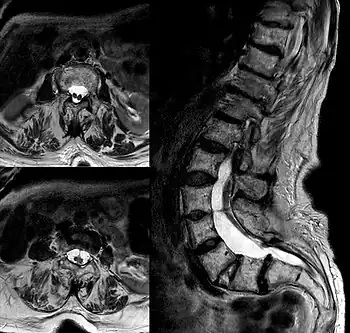 | |
| Diastematomelia in MRI of lumbar spine. | |
| Specialty | Medical genetics |
Signs and symptoms
The signs and symptoms of diastematomyelia may appear at any time of life, although the diagnosis is usually made in childhood. Cutaneous lesions (or stigmata), such as a hairy patch, dimple, Hemangioma, subcutaneous mass, Lipoma or Teratoma over the affected area of the spine is found in more than half of cases. Neurological symptoms are nonspecific, indistinguishable from other causes of cord tethering. The symptoms are caused by tissue attachments that limit the movement of the spinal cord within the spinal column. These attachments cause an abnormal stretching of the spinal cord.
The course of the disorder is progressive. In children, symptoms may include the "stigmata" mentioned above and/or foot and spinal deformities; weakness in the legs; low back pain; scoliosis; and incontinence. In adulthood, the signs and symptoms often include progressive sensory and motor problems and loss of bowel and bladder control. This delayed presentation of symptoms is related to the degree of strain placed on the spinal cord over time. Tethered spinal cord syndrome appears to be the result of improper growth of the neural tube during fetal development, and is closely linked to spina bifida.
Tethering may also develop after spinal cord injury and scar tissue can block the flow of fluids around the spinal cord. Fluid pressure may cause cysts to form in the spinal cord, a condition called syringomyelia. This can lead to additional loss of movement, feeling or the onset of pain or autonomic symptoms.
Cervical diastematomyelia can become symptomatic as a result of acute trauma, and can cause major neurological deficits, like hemiparesis, to result from otherwise mild trauma.[2]
The following definitions may help to understand some of the related entities:
- Diastematomyelia (di·a·stem·a·to·my·elia) is a congenital anomaly, often associated with spina bifida, in which the spinal cord is split into halves by a bony spicule or fibrous band, each half being surrounded by a dural sac.
- Myeloschisis (my·elos·chi·sis) is a developmental anomaly characterized by a cleft spinal cord, owing to failure of the neural plate to form a complete neural tube or to rupture of the neural tube after closure.
- Diplomyelia (diplo.my.elia) is a true duplication of spinal cord in which these are two dural sacs with two pairs of anterior and posterior nerve roots.
Pathophysiology
Diastematomyelia is a "dysraphic state" of unknown embryonic origin, but is probably initiated by an accessory neurenteric canal (an additional embryonic spinal canal.) This condition may be an isolated phenomenon or may be associated with other segmental anomalies of the vertebral bodies such as spina bifida, kyphoscoliosis, butterfly vertebra, hemivertebra and block vertebrae which are observed in most of the cases. Scoliosis is identified in more than half of these patients. In most of the symptomatic patients, the spinal cord is split into halves by a bony spicule or fibrous band, each half being surrounded by a dural sac. Other conditions, such as intramedullary tumors, tethered cord, dermoids, lipoma, syringomyelia, hydromyelia and Arnold–Chiari malformations have been described in medical literature, but they are exceptionally rare.
Diastematomyelia usually occurs between 9th thoracic and 1st sacral levels of the spinal column with most being at the level of the upper lumbar vertebra. Cervical diastematomyelia is a very rare entity. The extent (or length of spinal cord involved) varies from one affected individual to another. In approximately 60% of patients with diastematomyelia, the two hemicords, each covered by an intact layer of pia arachnoid, travel through a single subarachnoid space surrounded by a single dural sac. Each hemicord has its own anterior spinal artery. This form of diastematomyelia is not accompanied by any bony spur or fibrous band and is rarely symptomatic unless hydromyelia or tethering is present. The other 40% of patients have a bony spur or a fibrous band that passes through the two hemicords. In these cases, the dura and arachnoid are split into two separate dural and arachnoidal sacs, each surrounding the corresponding hemicord which are not necessarily symmetric. Each hemicord contains a central canal, one dorsal horn (giving rise to a dorsal nerve root), and one ventral horn (giving rise to a ventral nerve root.) One study showed the bony spur typically situated at the most inferior aspect of the dural cleft. They advised that if the imaging appears to show otherwise, a second spur (present in about 5% of patients with diastematomyelia) is likely to be present.The conus medullaris is situated below the L2 level in more than 75% of these diastematomyelia patients. Thickening of the filum terminale is seen in over half of the cases. While the level of the cleft is variable, it is most commonly found in the lumbar region. The two hemicords usually reunite caudally to the cleft. Occasionally, however, the cleft will extend unusually low and the cord will end with two separate coni medullarae and two fila terminale ("Diplomyelia").
Diagnosis
Adult presentation in diastematomyelia is unusual. With modern imaging techniques, various types of spinal dysraphism are being diagnosed in adults with increasing frequency. The commonest location of the lesion is at first to third lumbar vertebrae. Lumbosacral adult diastematomyelia is even rarer. Bony malformations and dysplasias are generally recognized on plain x-rays. MRI scanning is often the first choice of screening and diagnosis. MRI generally give adequate analysis of the spinal cord deformities although it has some limitations in giving detailed bone anatomy. Combined myelographic and post-myelographic CT scan is the most effective diagnostic tool in demonstrating the detailed bone, intradural and extradural pathological anatomy of the affected and adjacent spinal canal levels and of the bony spur.
Prenatal ultrasound diagnosis of this anomaly is usually possible in the early to mid third-trimester. An extra posterior echogenic focus between the fetal spinal laminae is seen with splaying of the posterior elements, thus allowing for early surgical intervention and have a favorable prognosis. Prenate ultrasound could also detect whether the diastematomyelia is isolated, with the skin intact or association with any serious neural tube defects. Progressive neurological lesions may result from the "tethering cord syndrome" (fixation of the spinal cord) by the diastematomyelia phenomenon or any of the associated disorders such as myelodysplasia, dysraphia of the spinal cord.
Treatment
Surgery
Surgical intervention is warranted in patients who present with new onset neurological signs and symptoms or have a history of progressive neurological manifestations that can be related to this abnormality. The surgical procedure required for the effective treatment of diastematomyelia includes decompression (surgery) of neural elements and removal of bony spur. This may be accomplished with or without resection and repair of the duplicated dural sacs. Resection and repair of the duplicated dural sacs is preferred since the dural abnormality may partly contribute to the "tethering" process responsible for the symptoms of this condition.
Post-myelographic CT scanning provides individualized detailed maps that enable surgical treatment of cervical diastematomyelia, first performed in 1983.[2][3]
Observation
Asymptomatic patients do not require surgical treatment. These patients should have regular neurological examinations since it is known that the condition can deteriorate. If any progression is identified, then a resection should be performed.
References
- Doherty, D.; Walker, W. O. (1 January 2014). "Neural Tube Defects". Encyclopedia of the Neurological Sciences (Second ed.). Academic Press. pp. 360–365. ISBN 978-0-12-385158-1. Retrieved 28 February 2023.
- Kuchner, E.F., Anand, A.K. & Kaufman, B.M., "Cervical Diastematomyelia" Neurosurgery, 16(4): 538-542, 1985
- Anand, A.K., Baim, R.S. & Kuchner, E.F., "Cervical Diastematomyelia" Computerized Radiology. 9(1):45-49, 1985