Discodermolide
(+)-Discodermolide is a polyketide natural product found to stabilize microtubules. (+)-discodermolide was isolated by Gunasekera and his co-workers at the Harbor Branch Oceanographic Institute from the deep-sea sponge Discodermia dissoluta in 1990.[1] (+)-Discodermolide was found to be a potent inhibitor of tumor cell growth in several MDR cancer cell lines. (+)-discodermolide also shows some unique characters, including a linear backbone structure, immunosuppressive properties both in vitro and in vivo,[2][3] potent induction of an accelerated senescence phenotype,[4] and synergistic antiproliferative activity in combination with paclitaxel.[5] Discodermolide was recognized as one of the most potent natural promoters of tubulin assembly. A large number of efforts toward the total synthesis of (+)-discodermolide were directed by its interesting biological activities and extreme scarcity of natural sources (0.002% w/w from frozen marine sponge). The compound supply necessary for complete clinical trials cannot be met by harvesting, isolation, and purification. As of 2005, attempts at synthesis or semi-synthesis by fermentation have proven unsuccessful. As a result, all discodermolide used in preclinical studies and clinical trials has come from large-scale total synthesis.[6]
 | |
| Names | |
|---|---|
| Systematic IUPAC name
(3Z,5S,6S,7S,8R,9S,11Z,13S,14S,15S,16Z,18S)-8,14,18-Trihydroxy-19-[(2S,3R,4S,5R)-4-hydroxy-3,5-dimethyl-6-oxooxan-2-yl]-5,7,9,11,13,15-hexamethylnonadeca-1,3,11,16-tetraen-6-yl carbamate | |
| Identifiers | |
3D model (JSmol) |
|
| ChEMBL | |
| ChemSpider | |
| KEGG | |
PubChem CID |
|
| UNII | |
CompTox Dashboard (EPA) |
|
| |
| |
| Properties | |
| C33H55NO8 | |
| Molar mass | 593.79 g/mol |
| Melting point | 112 to 113 °C (234 to 235 °F; 385 to 386 K) |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
Infobox references | |
History
Discodermolide was first isolated in 1990 from the Caribbean marine sponge Discodermia dissoluta by chemist Dr. Sarath Gunasekera and biologist Dr. Ross Longley, scientists at the Harbor Branch Oceanographic Institution.[1] The sponge contained 0.002% of discodermolide (7 mg/434 g of sponge). Since the compound is light-sensitive, the sponge must be harvested at a minimum depth of 33 meters. Discodermolide was initially found to have immunosuppressive and antifungal activities.
Structure
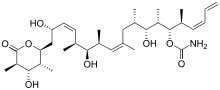
(+)-discodermolide has a linear polypropionate backbone, punctuated by Z-olefinic linkages at C(8,9) and C(13,14), a terminal Z-diene substituent at C(21–24), 13 stereogenic centers (including four secondary hydroxyls and seven methyl substituents), a carbamate, and a fully substituted D-lactone. The relative stereochemistry was determined by X-ray crystallography.[1] The absolute stereochemistry of (+)-discodermolide was reported by Schreiber and his co-workers in 1993.[7] Discodermolide adopts a U-shaped conformation, where the internal (Z)-alkenes act as conformational locks by minimizing allylic strain and syn-pentane interactions along the backbone. The D-lactone is held in a boat-like conformation.
Biological activities
Immunosuppressive properties
Initial biological evaluation of (+)-discodermolide by the Longley group showed that it has immunosuppressive properties both in vitro[2] and in vivo.[3] The immunosuppression response was observed at a relatively low concentration that (+)-discodermolide was non-toxic in vitro. In both human peripheral blood leukocytes and murine splenocytes, (+)-discodermolide was found to suppress the two-way mixed lymphocyte reaction. In addition, mitogenic response of peripheral blood leukocytes was also suppressed by the (+)-discodermolide. Follow up experiments demonstrated that (+)-discodermolide also has anti-proliferative effects in several other non-lymphoid cell lines.
Antiproliferative and antimitotic properties
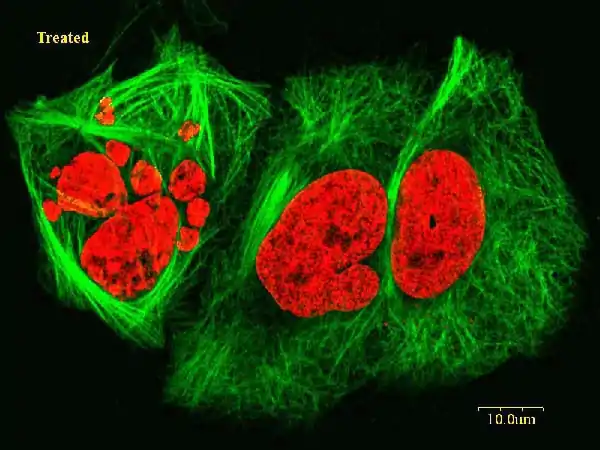
(+)-Discodermolide is a highly potent antiproliferative agent.[5] (+)-Discodermolide treated murine Do11.10T hybridoma cells could not proceed normal cell cycling. In untreated controls, 68% of cells were found at G1 phase, and 31% were found at S phase, and less than 1% was found at the G2/M phase. However, after 3 hours (+)-discodermolide treatment, 52% were found at G1 phase, 40% at S phase, and 58% at G2 and M phase. This result indicated that (+)-discodermolide blocks the cell cycle at G2 and M phase. This inhibition effect was also found to be reversible. Cells resume normal cycling within 48 hours after removal of (+)-discodermolide from the cell culture medium. (+)-Discodermolide arrests cell cycle via binding and stabilization of the microtubule network. Hyper-stabilization of the mitotic spindle causes cell cycle arresting at G2 and M phase and eventually leads to cell death by apoptosis. At 10µM concentration, (+)-discodermolide is able to promote microtubules assembly without microtubule-associated proteins and GTP. With microtubule-associated protein and GTP presented, 10µM (+)-discodermolide is able to initiate tubulin polymerization at 0 °C.[8][9]
Potent Inducer of accelerated cell senescence
(+)-Discodermolide is able to induce the senescence phenotype. (+)-Discodermolide treated Hela, MDA-MB-231, HCT-116, and A549 cells[4] exhibited moderated to high levels of β-galactosidase activity in all four cell lines. The β-galactosidase activity is one of the hallmarks of senescence.[10] Other common features include a cessation of proliferation and an increased cytoplasmic area. (+)-Discodermolide also induced up-regulation and activation in three proteins (p66Shc, Erk1, and Erk2).
Neuroprotective agent
(+)-Discodermolide has also been found to be neuroprotective in recent Alzheimer research.[11] The microtubule-stabilizing feature of (+)-discodermolide was used to restore neuron functions that have been disrupted by the amyloid induced sequestration. Tau protein is known to stabilize the microtubule network in healthy neurons.[12] It served as the “railroad track” upon which actin, tubulin, mitochondria, neurotransmitter-related enzymes and vesicles carrying messenger proteins are delivered. The presence of Amyloid in the cell could lead to tau protein aggregation and microtubule numbers reduction. In transgenic mouse model for human tauopathy, (+)-discodermolide stabilizes microtubules and restores fast axonal transport in cells, offsetting the loss of function caused by aggregation of tau protein. The motor-impaired mice exhibited a full restoration of normal movement with (+)-discodermolide treatment.
Potent natural promoter of tubulin assembly
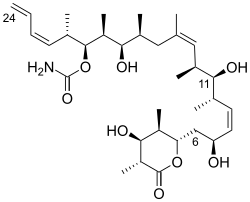
Discodermolide competes with paclitaxel[13] for microtubule binding, but with higher affinity and is also effective in paclitaxel- and in epothilone-resistant cancer cells. Discodermolide also seems to demonstrate a remarkably consistent 3D molecular conformation in the solid-state, in solution and when bound to tubulin; molecules with the conformational flexibility of discodermolide usually present very different conformations in different environments.
Biosynthesis
Many marine-derived polyketides that are often found in sponges cannot be cultured out of their natural environment. Finding the genes responsible for the biosynthesis of a sponge derived polyketides is a difficult task to accomplish because of the sponges’ colonial nature Scientists are not yet able to culture the sponges; therefore, the genes for the biosynthesis of (+)-discodermolide have not yet been discovered.[14]
Total syntheses
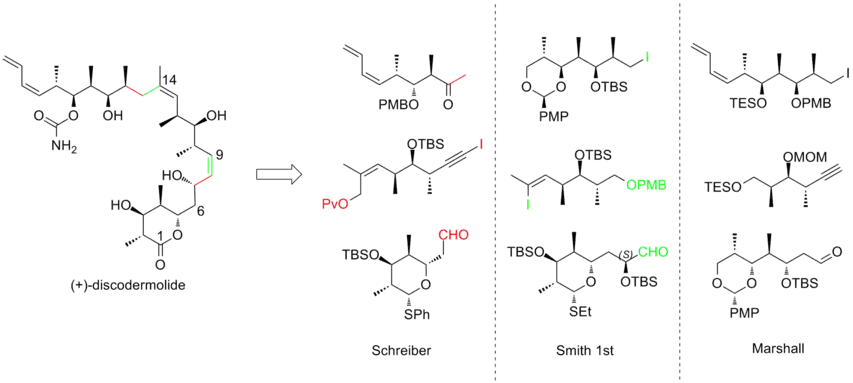
An intense effort has been made towards the total synthesis of (+)-discodermolide in order to meet the growing interest of studying its clinical profile. More than a dozen of syntheses have been published by different research groups. After decade of work, discodermolide syntheses have become more convergent and more practical. All of the total syntheses approaches started with the construction of three major fragments of roughly equivalent complexity, each of which contains the methyl-hydroxyl-methyl triad of contiguous centers that matches the stereogenicity of discodermolide target. Here are three examples of the retrosynthetic analyses of (+)-discodermolide:
The Schreiber synthesis of (-)-discodermolide
In 1993, Schreiber and his co-workers[7] reported the first total synthesis of the unnatural antipode (-)-discodermolide and determined the absolute stereochemistry of the natural product. The Schreiber team recognized three fragments of roughly equal complexity that are separated by olefinic units in discodermolide. The two starting materials, homoallylic alcohols, are both readily derived from 3-hydroxy-2-methylpropionate.
The trisubstituted (Z)-alkene of first fragment was generated by using the Still-Gennari reagent. Gilbert's reagent was then used to homologate it to an acetylene. Goekjian and Kishi method was then used to get the desired fragment, iodoacetylene. The same homoallylic alcohol was converted to diol in four steps. After oxidation to keto-aldehyde, it was homololated to dienes by a palladium-catalyzed coupling with vinylzinc bromide to generated the second fragment. The six-membered ring subunit was transformed from an acetal, which was obtained by an intramolecular 1,4-addition of a presumed hemiacetal intermediate.
-discodermolide_fragment.png.webp)
The Schreiber team found that the NiCl2 and CrCl2 promoted addition of alkynyl iodide to aldehyde effectively. The reaction provides a 2:1 mixture of desired product and its isomer. However, the undesired isomer can be recycled to the desired epimer in three steps, including Swern oxidation and Corey's asymmetric reduction. Pd catalyzed partial hydrogenation of the alkyne to establish the Z-configuration at C(8)–C(9). The pivaloyloxy group was then converted to a bromide for the final coupling. A stereoselective enolate alkylation between the allylic bromide with the lithium enolate derived from the second fragment provided a 3:1 mixture of isomers and completed the backbone assembly of (-)-discodermolide. During the final conversion, the NaBH4 reduction of ketone provided a separable 2.5:1 mixture of isomers. The Schreiber synthesis of (-)-discodermolide has an overall yield of 3.2% with a longest linear sequence of 24 steps and 36 total steps. In 1996, the Schreiber group published the total synthesis of natural discodermolide with similar synthetic strategy (4.3% overall yield, 36 steps, 24 steps longest linear sequence.
-discodermolide_endgame.png.webp)
The first-generation Smith synthesis of (-)-discodermolide
In 1995, Smith and his co-workers[15] reported the second total synthesis of (-)-discodermolide. Smith adapted the triply convergent strategy of the Schreiber synthesis. In Smith's strategy, all three fragments shared a common precursor, which was the product of a highly efficient 50g scale five-step conversion from 3-hydroxy-2-methylpropionate with 59% yield. An Evans syn aldol reaction was utilized to obtain the desired stereochemical outcome.
The synthesis of fragment A, alkyl iodide, applied the Evans' acyl oxazolidinone method. After hydroxyl protection and reductive removal of the chiral auxiliary, an iodination was carried out to afford fragment A. The preparation of fragment B began with TBS protection and DIBAL reduction. The resulting aldehyde can be converted to desired Z-trisubstituted vinyl halide with 6:1 selectivity. The key feature of the synthesis of fragment C was the addition of the anion derived from dithiane to benzyl glycidyl ether.
-discodermolide_fragment.png.webp)
Palladium(0)-mediated crosscoupling of vinyl iodide with the organozinc derivative of alkyl iodide afford product in 66% yield. After a two-step conversion to the corresponding phosphonium salt, Wittig union of phosphonium salt with aldehyde, fragment C, proceeded in 76% yield and good selectivity. The last feature of this synthesis was the titanium-mediated installation of the diene. The smith synthesis of (-)-discodermolide has an overall yield of 2.0% with a longest linear sequence of 29 steps and 42 total steps.
-discodermolide_endgame.png.webp)
The Myles synthesis of (-)-discodermolide
In 1997, Myles and his co-workers[16] has described the total synthesis of (-)-discodermolide using chelation-controlled alkylation as the key coupling. The titanium-mediated hetero-Diels–Alder reaction of aldehyde with the Danishefsky diene successfully produced the challenging Z-trisubstituted C(13)–C(14) olefin in allylic iodide fragment. The stereogenicity in the C(16)–C(21) subunit was induced by employing Evans aldol chemistry. The stereogenicity of the final fragment was set by a tin-promoted asymmetric allylation. Notably, the correct oxidation state at C(1) of this fragment would reduce the number of steps after the coupling of fragments. The Myles synthesis of (-)-discodermolide has an overall yield of 1.5% with a longest linear sequence of 25 steps and 44 total steps.
-discodermolide_endgame.png.webp)
The Marshall synthesis of (+)-discodermolide
Marshall and his co-workers[17] used their asymmetric allenylmetal-homoaldol tactic to construct polypropionate frameworks of (+)-discodermolide. The novelty of the Marshall approach is that the three stereotriad subunits are assembled through addition of non-racemic allenylmetal reagents to (S)-3-silyloxy-2-methylpropanal to generate both syn/syn and syn/anti adducts. The central feature for the synthesis of the alkyl iodide fragment was the treatment of aldehyde to allenyltributylstannane in the presence of BF3-etherate to get the syn/syn isomer. Syn/anti methyl-hydroxyl-methyl stereotriads were obtained through the reaction between aldehyde and homochiral allenylzinc reagent.
-discodermolide_fragment.png.webp)
In the Marshall synthesis, the coupling between the alkyne fragment to the aldehyde fragment proceeded in 92% yield, with 85:15 diastereoselectivity. The condensation of aldehyde with iodoethylidene triphenylphosphorane was the most challenging step, which produce 40% yield and an 85:15 inseparable mixture of (Z) and (E) isomers. The last coupling was performed via a Suzuki union of vinyl iodide with the boronate derived of alkyl iodide. The Marshall synthesis of (+)-discodermolide has an overall yield of 1.3% with a longest linear sequence of 30 steps and 48 total steps.
-discodermolide_endgame.png.webp)
The Evans synthesis of (+)-discodermolide
Evans and his co-workers[18] has developed a strategy that relies heavily on asymmetric aldol methodology for the production of the polypropionate backbone. They used a Claisen condensation reaction to construct the C(13)–C(14) trisubstituted Z-olefin. The Evans synthesis of (+)-discodermolide has an overall yield of 6.4% with a longest linear sequence of 31 steps and 49 total steps.
-discodermolide_fragment.png.webp)
-discodermolide_endgame.png.webp)
The Smith gram-scale synthesis of (+)-discodermolide
In 1999, Smith and his co-workers[19] reported the first gram-scale synthesis of (+)-discodermolide, which make the development of (+)-discodermolide as a potential chemotherapeutic agent possible. This synthetic route was licensed to Novartis Pharmaceuticals. One of the major improvements was that no purification was required in the first four steps of the five-step sequence towards the common precursor as the intermediate, aldol adduct, is a crystalline solid. A chelation-controlled Mukaiyama-aldol reaction was used to set the stereogenicity at C(5) of the lactone fragment.
-discodermolide_fragment.png.webp)
Two years later, Smith and his co-workers publish a third-generation synthesis in which the bulky TBS ether protecting group at C(11) of the precursor iodide was replaced by a less sterically encumbering group, MOM. This modification favors the formation of the desired phosphonium salt. The Smith third-generation synthesis of (+)-discodermolide has an overall yield of 6.0% with a longest linear sequence of 21 steps and 35 total steps.
-discodermolide_endgame.png.webp)
The fourth-generation Smith synthesis of (+)-discodermolide
The key feature of the fourth-generation[20] approach was the utilization of bidirectional fragment, vinyl iodide/phosphonium salt. phosphonium salt could be readily obtained as the trisubstituted vinyl iodide is less reactive than alkyl iodide. A Wittig reaction with followed by Suzuki coupling with alkyl iodide efficiently furnished the backbone of (+)-discodermolide. The Smith fourth-generation synthesis of (+)-discodermolide has an overall yield of 9.0% with a longest linear sequence of 17 steps and 36 total steps.
-discodermolide_endgame.png.webp)
The first-generation Paterson synthesis of (+)-discodermolide
Paterson and his co-workers[21] at the University of Cambridge have developed a strategy which utilizes novel chelation-controlled and reagent-controlled aldol reactions with high selectivity for subunit connections. Another feature of this synthesis was the induction of C(8)-C(9) olefin with cis geometry via Still-Gennari Horner-Wadsworth-Emmons reaction. The Paterson first-generation synthesis of (+)-discodermolide has an overall yield of 10.3% with a longest linear sequence of 23 steps and 42 total steps.
-discodermolide_endgame.png.webp)
The second- and third-generation Paterson syntheses of (+)-discodermolide
In 2003, Paterson and co-workers[22] reported a strategy that relies heavily on substrate-derived stereocontrol. Instead of a reagent-controlled aldol reaction in Paterson first-generation synthesis, a dicyclohexylboron-mediated anti-aldol was used to connect C(5)-C(6), which leads to a significant increase in diastereoselectivity from 4:1 to 92:8. Notably, Still–Gennari modified Horner–Wadsworth–Emmons reaction was also used to construct the C(13)-C(14) trisubstituted olefin in early stage of this synthesis. The Paterson second-generation synthesis of (+)-discodermolide has an overall yield of 7.8% with a longest linear sequence of 24 steps and 35 total steps.
-discodermolide_endgame.png.webp)
In 2004, Paterson and his co-workers[23] disclosed the third-generation total synthesis of (+)-discodermolide. The stepwise method used in previous generations to incorporate the C(1)–C(8) subunit was replaced by a late-stage Still-Gennari olefination, which leads to a notable improvement in convergence. The Paterson third-generation synthesis of (+)-discodermolide has an overall yield of 11.1% (highest reported to date) with a longest linear sequence of 21 steps and 37 total steps.
-discodermolide_endgame.png.webp)
The Novartis 60-g total synthesis of (+)-discodermolide
In early 2004, Novartis Pharmaceuticals[24] have disclosed the detail of a 60g-scale synthesis of (+)-discodermolide. This synthesis utilized the Smith gram-scale approach and the Paterson first-generation endgame. This synthesis allows (+)-discodermolide to be evaluated as an in vivo chemotherapeutic agent for adult patients presenting with advanced solid malignancies in Phase I clinical trials. The Novartis synthesis of (+)-discodermolide has an overall yield of 0.65% with a longest linear sequence of 26 steps and 33 total steps.
-discodermolide_endgame.png.webp)
The Panek total synthesis of (+)-discodermolide
In 2004, Panek and his co-workers[25] reported an approach which takes advantage of chiral crotylsilane-based C-C bond construction methodology to obtain the absolute stereochemistry of the three subunits of (+)-discodermolide. One of the key features of the Paterson approach is the utilization of hydrozirconation-cross-coupling methodology for the construction of C13-C14 (Z)-olefin. Acetate aldol reaction with high levels of 1,5-anti stereo induction and palladium-mediated sp2-sp3 cross coupling reaction are used to connect subunits. The Panek synthesis of (+)-discodermolide has an overall yield of 2.1% with a longest linear sequence of 27 steps and 42 total steps.
-discodermolide_endgame.png.webp)
The Ardisson total synthesis of (+)-discodermolide
In 2008, Ardisson and his co-workers[26] reported a strategy that applies a crotyltitanation reaction repeatedly to yield homoallylic (Z)-O-ene-carbamate alcohols with excellent selectivity. This crotyltitanation reaction not only efficiently produces the syn-anti methyl-hydroxy-methyl triads of (+)-discodermolide, but also yields products that can be easily converted to terminal (Z)-diene. The C13-C14 (Z)-olefin is installed through a highly selective dyotropic rearrangement. The Ardission synthesis of (+)-discodermolide has an overall yield of 1.6% with a longest linear sequence of 21 steps.
-discodermolide_endgame.png.webp)
Clinical development
The Harbor Branch Oceanographic Institution licensed (+)-discodermolide to Novartis, which began a phase 1 clinical trial in 2004. The trial concluded that the drug resulted in minimal toxicities and represents a novel mechanism of action.[27] Amos B. Smith's research group, in collaboration with Kosan Biosciences, has a preclinical drug development program ongoing.[28]
See also
References
- Gunasekera, S. P.; Gunasekera, M.; Longley, R. E.; Schulte, G. K. J. Org. Chem. 1990, 55, 4912-4915. (doi:10.1021/jo00303a029)
- Longley, R. E.; Caddigan, D.; Harmody, D.; Gunasekera, M.; Gunasekera, S. P. Transplantation 1991, 52, 650-655.
- Longley, R. E.; Caddigan, D.; Harmody, D.; Gunasekera, M.; Gunasekera, S. P. Transplantation 1991, 52, 656-661.
- Klein, L.; Freeze, B. S.; Smith, A. B., III; Horwitz, S. B. Cell Cycle 2005, 4, 501–507.
- Longley, R. E.; Gunasekera, S. P.; Faherty, D.; McLane, J.; Dumont, F. Ann. N.Y. Acad. Sci. 1993, 696, 94–107.
- Mickel, S. J. et al. Org. Process Res. Dev. 2004, 8
- Nerenberg, J. B.; Hung, D. T.; Somers, P. K.; Schreiber, S. L. J. Am. Chem. Soc. 1993, 115, 12621–12622
- ter Haar, E.; Kowalski, R. J.; Hamel, E.; Lin, C. M.; Longley, R. E.; Gunasekera, S. P.; Rosenkranz, H. S.; Day, B. W. Biochemistry 1996, 35, 243–250.
- Hung, D. T.; Chen, J.; Schreiber, S. L. Chem. Biol. 1996, 3, 287–293.
- Dimri, G.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; Peacocke, M.; Campisi, J. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 9363–9367.
- Forman, M. S.; Trojanowski, J. Q.; Lee, V. M.-Y. Nat. Med. 2004, 10, 1055–1063
- Zhang, B.; Maiti, A.; Shively, S.; Lakhani, F.; McDonald- Jones, G.; Bruce, J.; Lee, E. B.; Xie, S. X.; Joyce, S.; Li, C.; Toleikis, P. M.; Lee, V. M.-Y.; Trojanowski, J. Q. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 227–231.
- Martello, L. A.; McDaid, H. M.; Regl, D. L.; Yang, C.-P. H.; Meng, D.; Pettus, T. R. R.; Kaufman, M. D.; Arimoto, H.; Danishefsky, S. J.; Smith, A. B., III; Horwitz, S. B. Clin. Cancer Res. 2000, 6, 1978–1987
- Shaw, S. J.; Zhang, D.; Sundermann, K. F.; Myles, D. C. Fragment Assembly: An Alternative Approach to Generating Complex Polyketides. Synthetic Commun. 2005, 35, 1735-1743.
- Smith, A. B., III; Qiu, Y.; Jones, D. R.; Kobayashi, K. J. Am. Chem. Soc. 1995, 117, 12011–12012
- Harried, S. S.; Yang, G.; Strawn, M. A.; Myles, D. C. J. Org. Chem. 1997, 62, 6098–6099
- Marshall, J. A.; Johns, B. A. J. Org. Chem. 1998, 63, 7885–7892
- Halstead, D. P. Ph.D. Thesis, Harvard University, Cambridge, MA, 1999
- Smith, A. B., III; Kaufman, M. D.; Beauchamp, T. J.; LaMarche, M. J.; Arimoto, H. Org. Lett. 1999, 1, 1823–1826
- Smith, A. B., III; Freeze, B. S.; Xian, M.; Hirose, T. Org. Lett. 2005, 7, 1825–1828
- Paterson, I.; Florence, G. J.; Gerlach, K.; Scott, J. Angew. Chem. Int. Ed. 2000, 39, 377–380
- Paterson, I.; Delgado, O.; Florence, G. J.; Lyothier, I.; Scott, J. P.; Sereinig, N. Org. Lett. 2003, 5, 35–38
- Paterson, I.; Lyothier, I. Org. Lett. 2004, 6, 4933–4936
- Mickel, S. J.; Niederer, D.; Daeffler, R.; Osmani, A.; Kuesters, E.; Schmid, E.; Schaer, K.; Gamboni, R.; Chen, W.; Loeser, E.; Kinder, F. R., Jr.; Konigsberger, K.; Prasad, K.; Ramsey, T. M.; Repic, O.; Wang, R.-M.; Florence, G.; Lyothier, I.; Paterson, I. Org. Process Res. Dev. 2004, 8, 122–130
- Arefolov, A.; Panek, J. S. J. Am. Chem. Soc. 2005, 127, 5596–5603
- de Lemos, Elsa; Poree, Francois-Hugues; Bourin, Arnaud; Barbion, Julien; Agouridas, Evangelos; Lannou, Marie-Isabelle; Commercon, Alain; Betzer, Jean-Francois; Pancrazi, Ange; Ardisson, Janick. Chemistry - A European Journal (2008), 14(35), 11092-11112
- A phase I pharmacokinetic (PK) trial of XAA296A (Discodermolide) administered every 3 wks to adult patients with advanced solid malignancies. 2004 ASCO Annual Meeting ()
- Amos B. Smith, III Current Research Projects
External links
- Chemical and Engineering News: Scaled-Up Synthesis of Discodermolide by Michael Freemantle
- Chemistry and Biology of Discodermolide
- The Betzer and Ardisson Synthesis of (+)-Discodermolide