Disease gene identification
Disease gene identification is a process by which scientists identify the mutant genotypes responsible for an inherited genetic disorder. Mutations in these genes can include single nucleotide substitutions, single nucleotide additions/deletions, deletion of the entire gene, and other genetic abnormalities.
Significance
Knowledge of which genes (when non-functional) cause which disorders will simplify diagnosis of patients and provide insights into the functional characteristics of the mutation. The advent of modern-day high-throughput sequencing technologies combined with insights provided from the growing field of genomics is resulting in more rapid disease gene identification, thus allowing scientists to identify more complex mutations.
Generic gene identification procedure
Disease gene identification techniques often follow the same overall procedure. DNA is first collected from several patients who are believed to have the same genetic disease. Then, their DNA samples are analyzed and screened to determine probable regions where the mutation could potentially reside. These techniques are mentioned below. These probable regions are then lined-up with one another and the overlapping region should contain the mutant gene. If enough of the genome sequence is known, that region is searched for candidate genes. Coding regions of these genes are then sequenced until a mutation is discovered or another patient is discovered, in which case the analysis can be repeated, potentially narrowing down the region of interest.
The differences between most disease gene identification procedures are in the second step (where DNA samples are analyzed and screened to determine regions in which the mutation could reside).
Pre-genomics techniques
Without the aid of the whole-genome sequences, pre-genomics investigations looked at select regions of the genome, often with only minimal knowledge of the gene sequences they were looking at. Genetic techniques capable of providing this sort of information include Restriction Fragment Length Polymorphism (RFLP) analysis and microsatellite analysis.
Loss of heterozygosity (LOH)
Loss of heterozygosity (LOH) is a technique that can only be used to compare two samples from the same individual. LOH analysis is often used when identifying cancer-causing oncogenes in that one sample consists of (mutant) tumor DNA and the other (control) sample consists of genomic DNA from non-cancerous cells from the same individual. RFLPs and microsatellite markers provide patterns of DNA polymorphisms, which can be interpreted as residing in a heterozygous region or a homozygous region of the genome. Provided that all individuals are affected with the same disease resulting from a manifestation of a deletion of a single copy of the same gene, all individuals will contain one region where their control sample is heterozygous but the mutant sample is homozygous - this region will contain the disease gene.[1][2]
Post-genomics techniques
With the advent of modern laboratory techniques such as High-throughput sequencing and software capable of genome-wide analysis, sequence acquisition has become increasingly less expensive and time-consuming, thus providing significant benefits to science in the form of more efficient disease gene identification techniques.
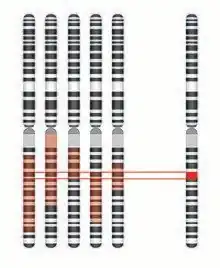
Identity by descent mapping
Identity by descent (IBD) mapping generally uses single nucleotide polymorphism (SNP) arrays to survey known polymorphic sites throughout the genome of affected individuals and their parents and/or siblings, both affected and unaffected. While these SNPs probably do not cause the disease, they provide valuable insight into the makeup of the genomes in question. A region of the genome is considered identical by descent if contiguous SNPs share the same genotype. When comparing an affected individual to his/her affected sibling, all identical regions are recorded (ex. Shaded in red in above figure). Given that an affected sibling and an unaffected sibling do not have the same disease phenotype, their DNA must by definition be different (barring the presence of a genetic or environmental modifier). Thus, the IBD mapping results can be further supplemented by removing any regions that are identical in both affected individuals and unaffected siblings.[3] This is then repeated for multiple families, thus generating a small, overlapping fragment, which theoretically contains the disease gene.
Homozygosity/autozygosity mapping
Homozygosity/Autozygosity mapping is a powerful technique, but is only valid when searching for a mutation segregating within a small, closed population. Such a small population, possibly created by the founder effect, will have a limited gene pool, and thus any inherited disease will probably be a result of two copies of the same mutation segregating on the same haplotype. Since affected individuals will probably be homozygous in the regions, looking at SNPs in a region is an adequate marker of regions of homozygosity and heterozygosity. Modern day SNP arrays are used to survey the genome and identify large regions of homozygosity. Homozygous blocks in the genomes of affected individuals can then be laid on top of each other, and the overlapping region should contain the disease gene.[4]
This analysis is often extended by analyzing autozygosity, an extension of homozygosity, in the genomes of affected individuals.[5] This can be accomplished by plotting a cumulative LOD score alongside the overlaid blocks of homozygosity. By taking into consideration the population allele frequencies for all SNPs via autozygosity mapping, the results of homozygosity can be confirmed.[5] Furthermore, if two suspicious regions appear as a result of homozygosity mapping, autozygosity mapping may be able to distinguish between the two (ex. If one block of homozygosity is a result of a very non-diverse region of the genome, the LOD score will be very low).
Tools for Homozygosity Mapping
- HomSI: a homozygous stretch identifier from next-generation sequencing data[6] A tool that identifies homozygous regions using deep sequence data.
Genome-wide knockdown studies
Genome-wide knockdown studies are an example of the reverse genetics made possible by the acquisition of whole genome sequences, and the advent of genomics and gene-silencing technologies, mainly siRNA and deletion mapping. Genome-wide knockdown studies involve systematic knockdown or deletion of genes or segments of the genome.[7] This is generally done in prokaryotes or in a tissue culture environment due to the massive number of knockdowns that must be performed.[8] After the systematic knockout is completed (and possibly confirmed by mRNA expression analysis), the phenotypic results of the knockdown/knockout can be observed. Observation parameters can be selected to target a highly specific phenotype.[8] The resulting dataset is then queried for samples which exhibit phenotypes matching the disease in question – the gene(s) knocked down/out in said samples can then be considered candidate disease genes for the individual in question.
Whole exome sequencing
Whole exome sequencing is a brute-force approach that involves using modern day sequencing technology and DNA sequence assembly tools to piece together all coding portions of the genome. The sequence is then compared to a reference genome and any differences are noted. After filtering out all known benign polymorphisms, synonymous changes, and intronic changes (that do not affect splice sites), only potentially pathogenic variants will be left. This technique can be combined with other techniques to further exclude potentially pathogenic variants should more than one be identified.[9]
References
- Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF, Nakamura Y, White R, Vogelstein B (April 1989). "Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas". Science. 244 (4901): 217–21. Bibcode:1989Sci...244..217B. doi:10.1126/science.2649981. PMID 2649981.
- Lee AS, Seo YC, Chang A, Tohari S, Eu KW, Seow-Choen F, McGee JO (September 2000). "Detailed deletion mapping at chromosome 11q23 in colorectal carcinoma". Br. J. Cancer. 83 (6): 750–5. doi:10.1054/bjoc.2000.1366. PMC 2363538. PMID 10952779.
- Bell R, Herring SM, Gokul N, Monita M, Grove ML, Boerwinkle E, Doris PA (June 2011). "High-resolution identity by descent mapping uncovers the genetic basis for blood pressure differences between spontaneously hypertensive rat lines". Circ Cardiovasc Genet. 4 (3): 223–31. doi:10.1161/CIRCGENETICS.110.958934. PMC 3116070. PMID 21406686.
- Sherman EA, Strauss KA, Tortorelli S, Bennett MJ, Knerr I, Morton DH, Puffenberger EG (November 2008). "Genetic mapping of glutaric aciduria, type 3, to chromosome 7 and identification of mutations in c7orf10". Am. J. Hum. Genet. 83 (5): 604–9. doi:10.1016/j.ajhg.2008.09.018. PMC 2668038. PMID 18926513.
- Broman KW, Weber JL (December 1999). "Long homozygous chromosomal segments in reference families from the centre d'Etude du polymorphisme humain". Am. J. Hum. Genet. 65 (6): 1493–500. doi:10.1086/302661. PMC 1288359. PMID 10577902.
- Zeliha Görmez; Burcu Bakir-Gungor; Mahmut Şamil Sağıroğlu (Feb 2014). "HomSI: a homozygous stretch identifier from next-generation sequencing data". Bioinformatics. 30 (3): 445–447. doi:10.1093/bioinformatics/btt686. PMID 24307702.
- Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ (May 2009). "A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene". Cell. 137 (5): 835–48. doi:10.1016/j.cell.2009.05.006. PMC 2768667. PMID 19490893.
- Fortier S, Bilodeau M, Macrae T, Laverdure JP, Azcoitia V, Girard S, Chagraoui J, Ringuette N, Hébert J, Krosl J, Mayotte N, Sauvageau G (2010). "Genome-wide interrogation of Mammalian stem cell fate determinants by nested chromosome deletions". PLOS Genet. 6 (12): e1001241. doi:10.1371/journal.pgen.1001241. PMC 3000362. PMID 21170304.
- Chen WJ, Lin Y, Xiong ZQ, Wei W, Ni W, Tan GH, Guo SL, He J, Chen YF, Zhang QJ, Li HF, Lin Y, Murong SX, Xu J, Wang N, Wu ZY (December 2011). "Exome sequencing identifies truncating mutations in PRRT2 that cause paroxysmal kinesigenic dyskinesia". Nat. Genet. 43 (12): 1252–5. doi:10.1038/ng.1008. PMID 22101681. S2CID 16129198.
- Johnson GC, Esposito L, Barratt BJ, Smith AN, Heward J, Di Genova G, Ueda H, Cordell HJ, Eaves IA, Dudbridge F, Twells RC, Payne F, Hughes W, Nutland S, Stevens H, Carr P, Tuomilehto-Wolf E, Tuomilehto J, Gough SC, Clayton DG, Todd JA (2001). "Haplotype tagging for the identification of common disease genes". Nat Genet. 29 (2): 233–7. doi:10.1038/ng1001-233. PMID 11586306. S2CID 3388593.