Unfolded protein response
The unfolded protein response (UPR) is a cellular stress response related to the endoplasmic reticulum (ER) stress.[1] It has been found to be conserved between mammalian species,[2] as well as yeast[1][3] and worm organisms.
The UPR is activated in response to an accumulation of unfolded or misfolded proteins in the lumen of the endoplasmic reticulum. In this scenario, the UPR has three aims: initially to restore normal function of the cell by halting protein translation, degrading misfolded proteins, and activating the signalling pathways that lead to increasing the production of molecular chaperones involved in protein folding. If these objectives are not achieved within a certain time span or the disruption is prolonged, the UPR aims towards apoptosis.
Sustained overactivation of the UPR has been implicated in prion diseases as well as several other neurodegenerative diseases, and inhibiting the UPR could become a treatment for those diseases.[4] Diseases amenable to UPR inhibition include Creutzfeldt–Jakob disease, Alzheimer's disease, Parkinson's disease, and Huntington's disease.[5]
Protein folding in the endoplasmic reticulum
Protein synthesis
The term protein folding incorporates all the processes involved in the production of a protein after the nascent polypeptides have become synthesized by the ribosomes. The proteins destined to be secreted or sorted to other cell organelles carry an N-terminal signal sequence that will interact with a signal recognition particle (SRP). The SRP will lead the whole complex (Ribosome, RNA, polypeptide) to the ER membrane. Once the sequence has “docked”, the protein continues translation, with the resultant strand being fed through the polypeptide translocator directly into the ER. Protein folding commences as soon as the polypeptide enters to the luminal environment, even as translation of the remaining polypeptide continues.
Protein folding and quality control
Protein folding steps involve a range of enzymes and molecular chaperones to coordinate and regulate reactions, in addition to a range of substrates required in order for the reactions to take place. The most important of these to note are N-linked glycosylation and disulfide bond formation. N-linked glycosylation occurs as soon as the protein sequence passes into the ER through the translocon, where it is glycosylated with a sugar molecule that forms the key ligand for the lectin molecules calreticulin (CRT; soluble in ER lumen) and calnexin (CNX; membrane bound).[6] Favoured by the highly oxidizing environment of the ER, protein disulfide isomerases facilitate formation of disulfide bonds, which confer structural stability to the protein in order for it to withstand adverse conditions such as extremes of pH and degradative enzymes.
The ER is capable of recognizing misfolding proteins without causing disruption to the functioning of the ER. The aforementioned sugar molecule remains the means by which the cell monitors protein folding, as the misfolding protein becomes characteristically devoid of glucose residues, targeting it for identification and re-glycosylation by the enzyme UGGT (UDP-glucose:glycoprotein glucosyltransferase).[6] If this fails to restore the normal folding process, exposed hydrophobic residues of the misfolded protein are bound by the protein glucose regulate protein 78 (Grp78), a heat shock protein 70kDa family member[7] that prevents the protein from further transit and secretion.[8]
Where circumstances continue to cause a particular protein to misfold, the protein is recognized as posing a threat to the proper functioning of the ER, as they can aggregate to one another and accumulate. In such circumstances the protein is guided through endoplasmic reticulum-associated degradation (ERAD). The chaperone EDEM guides the retrotranslocation of the misfolded protein back into the cytosol in transient complexes with PDI and Grp78.[9] Here it enters the ubiquitin-proteasome pathway, as it is tagged by multiple ubiquitin molecules, targeting it for degradation by cytosolic proteasomes.
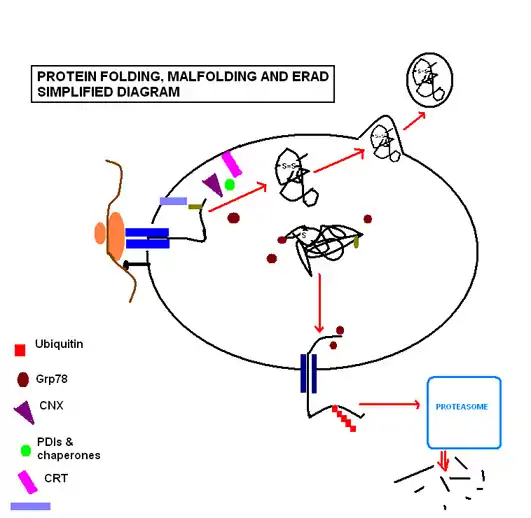
Successful protein folding requires a tightly controlled environment of substrates that include glucose to meet the metabolic energy requirements of the functioning molecular chaperones; calcium that is stored bound to resident molecular chaperones; and redox buffers that maintain the oxidizing environment required for disulfide bond formation.[10]
Unsuccessful protein folding can be caused by HLA-B27, disturbing balance of important (IL-10 and TNF) signaling proteins. At least some disturbances are reliant on correct HLA-B27 folding.[11]
However, where circumstances cause a more global disruption to protein folding that overwhelms the ER's coping mechanisms, the UPR is activated.
Molecular mechanism
Initiation
The molecular chaperone BiP/Grp78 has a range of functions within the ER. It maintains specific transmembrane receptor proteins involved in initiation of the downstream signalling of the UPR in an inactive state by binding to their luminal domains. An overwhelming load of misfolded proteins or simply the over-expression of proteins (e.g. IgG)[12] requires more of the available BiP/Grp78 to bind to the exposed hydrophobic regions of these proteins, and consequently BiP/Grp78 dissociates from these receptor sites to meet this requirement. Dissociation from the intracellular receptor domains allows them to become active. PERK dimerizes with BiP in resting cells and oligomerizes in ER-stressed cells.
Although this is traditionally the accepted model, doubts have been raised over its validity. It has been argued that the genetic and structural evidence supporting the model simply shows BiP dissociation to be merely correlated with Ire1 activation, rather than specifically causing it.[13] An alternative model has been proposed, whereby unfolded proteins interact directly with the ER-lumenal domain of Ire1, causing oligomerization and transautophosphorylation.[13] However these models are not mutually exclusive, it is also possible that both direct interaction of Ire1 with unfolded proteins and dissosiation of BiP from IRE1 contribute to the activation of the Ire1 pathway.
Functions
The initial phases of UPR activation have two key roles:
Translation Attenuation and Cell Cycle Arrest by the PERK Receptor This occurs within minutes to hours of UPR activation to prevent further translational loading of the ER. PERK (protein kinase RNA-like endoplasmic reticulum kinase) activates itself by oligomerization and autophosphorylation of the free luminal domain. The activated cytosolic domain causes translational attenuation by directly phosphorylating the α subunit of the regulating initiator of the mRNA translation machinery, eIF2.[14] This also produces translational attenuation of the protein machinery involved in running the cell cycle, producing cell cycle arrest in the G1 phase.[15] PERK deficiency may have a significant impact on physiological states associated with ER stress.
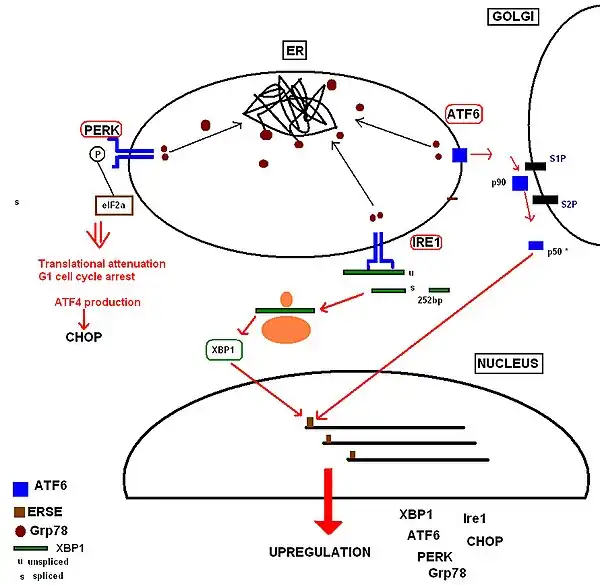
Increased Production of Proteins Involved in the Functions of the UPR UPR activation also results in upregulation of proteins involved in chaperoning malfolding proteins, protein folding and ERAD, including further production of Grp78. Ultimately this increases the cell's molecular mechanisms by which it can deal with the misfolded protein load. These receptor proteins have been identified as:
- Inositol-requiring kinase 1,[16] whose free luminal domain activates itself by homodimerisation and transautophosphorylation.[17] The activated domain is able to activate the transcription factor XBP1(Xbox binding protein) mRNA (the mammalian equivalent of the yeast Hac1 mRNA) by cleavage and removal of a 26bp intron. The activated transcription factor upregulates UPR 'stress genes' by directly binding to stress element promoters in the nucleus.[18]
- ATF6 (activating transcription factor 6) is a basic leucine zipper transcription factor.[19] Upon Grp78 dissociation, the entire 90kDa protein translocates to the Golgi, where it is cleaved by proteases to form an active 50kDa transcription factor[20] that translocates to the nucleus. It binds to stress element promoters upstream of genes that are upregulated in the UPR.[21]
The aim of these responses is to remove the accumulated protein load whilst preventing any further addition to the stress, so that normal function of the ER can be restored as soon as possible.
If the UPR pathway is activated in an abnormal fashion, such as when obesity triggers chronic ER stress and the pathway is constitutively active, this can lead to insensitivity to insulin signaling and thus insulin resistance. Individuals suffering from obesity have an elevated demand placed on the secretory and synthesis systems of their cells. This activates cellular stress signaling and inflammatory pathways because of the abnormal conditions disrupting ER homeostasis.
A downstream effect of the ER stress is a significant decrease in insulin-stimulated phosphorylation of tyrosine residues of insulin receptor substrate (IRS-1), which is the substrate for insulin tyrosine kinase (the insulin receptor). C-Jun N-terminal kinase (JNK) is also activated at high levels by IRE-1α, which itself is phosphorylated to become activated in the presence of ER stress. Subsequently, JNK phosphorylates serine residues of IRS-1, and thus inhibits insulin receptor signaling. IRE-1α also recruits tumor necrosis factor receptor-associated factor 2 (TRAF2). This kinase cascade that is dependent on IRE-1α and JNK mediates ER stress–induced inhibition of insulin action.[22]
Obesity provides chronic cellular stimuli for the UPR pathway as a result of the stresses and strains placed upon the ER, and without allowing restoration to normal cellular responsiveness to insulin hormone signaling, an individual becomes very likely to develop type 2 diabetes.
Skeletal muscles are sensitive to physiological stress, as exercise can impair ER homeostasis. This causes the expression of ER chaperones to be induced by the UPR in response to the exercise-induced ER stress. Muscular contraction during exercise causes calcium to be released from the sarcoplasmic reticulum (SR), a specialized ER network in skeletal muscles. This calcium then interacts with calcineurin and calcium/calmodulin-dependent kinases that in turn activate transcription factors. These transcription factors then proceed to alter the expression of exercise-regulated muscle genes. PGC-1alpha, a transcriptional coactivator, is a key transcription factor involved in mediating the UPR in a tissue-specific manner in skeletal muscles by coactivating ATF6alpha. Therefore, PGC-1alpha gets expressed in muscles after acute and long-term exercise training. The function of this transcription factor is to increase the number and function of mitochondria, as well as to induce a switch of skeletal fibers to slow oxidative muscle fibers, as these are fatigue-resistant. Therefore, this UPR pathway mediates changes in muscles that have undergone endurance training by making them more resistant to fatigue and protecting them from future stress.[23]
Initiating apoptosis
In conditions of prolonged stress, the goal of the UPR changes from being one that promotes cellular survival to one that commits the cell to a pathway of apoptosis. Proteins downstream of all 3 UPR receptor pathways have been identified as having pro-apoptotic roles. However, the point at which the 'apoptotic switch' is activated has not yet been determined, but it is a logical consideration that this should be beyond a certain time period in which resolution of the stress has not been achieved. The two principal UPR receptors involved are Ire1 and PERK.
By binding with the protein TRAF2, Ire1 activates a JNK signaling pathway,[24] at which point human procaspase 4 is believed to cause apoptosis by activating downstream caspases.
Although PERK is recognised to produce a translational block, certain genes can bypass this block. An important example is that the proapoptotic protein CHOP (CCAAT/-enhancer-binding protein homologous protein), is upregulated downstream of the bZIP transcription factor ATF4 (activating transcription factor 4) and uniquely responsive to ER stress.[25] CHOP causes downregulation of the anti-apoptotic mitochondrial protein Bcl-2,[26] favouring a pro-apoptotic drive at the mitochondria by proteins that cause mitochondrial damage, cytochrome c release and caspase 3 activation.
Diseases
Diseases amenable to UPR inhibition include Creutzfeldt–Jakob disease, Alzheimer's disease, Parkinson's disease, and Huntington's disease.[5]
Endoplasmic reticulum stress was reported to play a major role in non‐alcoholic fatty liver disease (NAFLD) induction and progression. High fat diet fed rats showed increased ER stress markers CHOP, XBP1, and GRP78. ER stress is known to activate hepatic de novo lipogenesis, inhibit VLDL secretion, promote insulin resistance and inflammatory process, and promote cell apoptosis. Thus it increase the level of fat accumulation and worsens the NAFLD to a more serious hepatic state.[27] Zingiber officinale (ginger) extract and omega‐3 fatty acids were reported to ameliorate endoplasmic reticulum stress in a nonalcoholic fatty liver rat model.[27]
As stated above, the UPR can also be activated as a compensatory mechanism in disease states. For instance, the UPR is up-regulated in an inherited form of dilated cardiomyopathy due to a mutation in gene encoding the Phospholamban protein.[28] Further activation proved therapeutic in a human induced pluripotent stem cell model of PLN mutant dilated cardiomyopathy.[28]
Chemical inducers
- Brefeldin A is a very common inducer of the unfolded protein response or endoplasmic reticulum stress response (ER stress).
- thapsigargin[29] leads to ER Ca2+ depletion due to inhibition of the Sarco/Endoplasmic Reticulum Ca2+-ATPase (SERCA).
- A23187[29] upregulates expression of ER stress proteins
- 2-deoxyglucose[29]
- dithiothreitol[29] reduces the disulfide bridges of proteins. The denatured proteins accumulated inside the ER.
- fenretinide and bortezomib (Velcade), each acting via different cellular mechanisms, induce ER stress, leading to apoptosis in melanoma cells.
- tunicamycin inhibits N-linked glycosylation.
Biological inducers
- Dengue virus induces PERK dependent ER stress as part of virus induced response in infected cells to favor replication.[30]
- Influenza virus requires endoplasmic reticulum protein 57-kD (ERp57) for replication and apoptosis induction in infected cells.[31]
See also
References
- Hetz C, Papa FR (January 2018). "The Unfolded Protein Response and Cell Fate Control". Molecular Cell. 69 (2): 169–181. doi:10.1016/j.molcel.2017.06.017. PMID 29107536.
- "Peter Walter's short talk: Unfolding the UPR". Archived from the original on 2017-07-12. Retrieved 2013-10-24.
- Kannan M, Sivaprakasam C, Prinz WA, Nachiappan V (December 2016). "Endoplasmic reticulum stress affects the transport of phosphatidylethanolamine from mitochondria to the endoplasmic reticulum in S.cerevisiae". Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1861 (12 Pt A): 1959–1967. doi:10.1016/j.bbalip.2016.09.015. PMC 6322925. PMID 27678054.
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, et al. (October 2013). "Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice". Science Translational Medicine. 5 (206): 206ra138. doi:10.1126/scitranslmed.3006767. PMID 24107777. S2CID 25570626.
- BBC Health News (2013-10-10). "Alzheimer's breakthrough hailed as 'turning point'". British Broadcasting Co. Retrieved 2013-10-10.
- Blond-Elguindi S, Cwirla SE, Dower WJ, Lipshutz RJ, Sprang SR, Sambrook JF, Gething MJ (November 1993). "Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP". Cell. 75 (4): 717–28. doi:10.1016/0092-8674(93)90492-9. PMID 7902213.
- Brewer JW, Diehl JA (November 2000). "PERK mediates cell-cycle exit during the mammalian unfolded protein response". Proceedings of the National Academy of Sciences of the United States of America. 97 (23): 12625–30. Bibcode:2000PNAS...9712625B. doi:10.1073/pnas.220247197. PMC 18814. PMID 11035797.
- Chen X, Shen J, Prywes R (April 2002). "The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi". The Journal of Biological Chemistry. 277 (15): 13045–52. doi:10.1074/jbc.M110636200. PMID 11821395.
- Cox JS, Shamu CE, Walter P (June 1993). "Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase". Cell. 73 (6): 1197–206. doi:10.1016/0092-8674(93)90648-A. PMID 8513503. S2CID 16065404.
- Hammond C, Braakman I, Helenius A (February 1994). "Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control". Proceedings of the National Academy of Sciences of the United States of America. 91 (3): 913–7. Bibcode:1994PNAS...91..913H. doi:10.1073/pnas.91.3.913. PMC 521423. PMID 8302866.
- LL Markus Penttinen (January 10, 2004). HLA-B27 associated with debilitated salmonella bacteria resistance (in Finnish). Turku University Library: Ann. Univ. Turkuensis D 619. ISBN 951-29-2742-X. Archived from the original on January 6, 2013. Retrieved October 9, 2012.
- Kober L, Zehe C, Bode J (October 2012). "Development of a novel ER stress based selection system for the isolation of highly productive clones". Biotechnology and Bioengineering. 109 (10): 2599–611. doi:10.1002/bit.24527. PMID 22510960. S2CID 25858120.
- Bernales S, Papa FR, Walter P (2006). "Intracellular signaling by the unfolded protein response". Annual Review of Cell and Developmental Biology. 22: 487–508. doi:10.1146/annurev.cellbio.21.122303.120200. PMID 16822172.
- Harding HP, Zhang Y, Ron D (January 1999). "Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase". Nature. 397 (6716): 271–4. Bibcode:1999Natur.397..271H. doi:10.1038/16729. PMID 9930704. S2CID 4416662.
- Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH (August 2003). "Proteasome inhibitors disrupt the unfolded protein response in myeloma cells". Proceedings of the National Academy of Sciences of the United States of America. 100 (17): 9946–51. Bibcode:2003PNAS..100.9946L. doi:10.1073/pnas.1334037100. PMC 187896. PMID 12902539.
- Lee AS (January 1987). "Coordinated regulation of a set of genes by glucose and calcium ionophores in mammalian cells". Trends in Biochemical Sciences. 12: 20–3. doi:10.1016/0968-0004(87)90011-9.
- Machamer CE, Doms RW, Bole DG, Helenius A, Rose JK (April 1990). "Heavy chain binding protein recognizes incompletely disulfide-bonded forms of vesicular stomatitis virus G protein". The Journal of Biological Chemistry. 265 (12): 6879–83. doi:10.1016/S0021-9258(19)39231-2. PMID 2157712.
- Stĕrba O (1975). "Prenatal growth of the mole, Talpa europaea Linn., 1758". Folia Morphologica. 23 (3): 282–5. PMID 1158311.
- Molinari M, Galli C, Piccaluga V, Pieren M, Paganetti P (July 2002). "Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER". The Journal of Cell Biology. 158 (2): 247–57. doi:10.1083/jcb.200204122. PMC 2173128. PMID 12119363.
- Mori K, Ogawa N, Kawahara T, Yanagi H, Yura T (April 2000). "mRNA splicing-mediated C-terminal replacement of transcription factor Hac1p is required for efficient activation of the unfolded protein response". Proceedings of the National Academy of Sciences of the United States of America. 97 (9): 4660–5. Bibcode:2000PNAS...97.4660M. doi:10.1073/pnas.050010197. PMC 18289. PMID 10781071.
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D (January 2000). "Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1". Science. 287 (5453): 664–6. Bibcode:2000Sci...287..664U. doi:10.1126/science.287.5453.664. PMID 10650002.
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. (October 2004). "Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes". Science. 306 (5695): 457–61. Bibcode:2004Sci...306..457O. doi:10.1126/science.1103160. PMID 15486293. S2CID 22517395.
- Wu J, Ruas JL, Estall JL, Rasbach KA, Choi JH, Ye L, et al. (February 2011). "The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1α/ATF6α complex". Cell Metabolism. 13 (2): 160–9. doi:10.1016/j.cmet.2011.01.003. PMC 3057411. PMID 21284983.
- Wang XZ, Lawson B, Brewer JW, Zinszner H, Sanjay A, Mi LJ, Boorstein R, Kreibich G, Hendershot LM, Ron D (August 1996). "Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153)". Molecular and Cellular Biology. 16 (8): 4273–80. doi:10.1128/mcb.16.8.4273. PMC 231426. PMID 8754828.
- Welihinda AA, Kaufman RJ (July 1996). "The unfolded protein response pathway in Saccharomyces cerevisiae. Oligomerization and trans-phosphorylation of Ire1p (Ern1p) are required for kinase activation". The Journal of Biological Chemistry. 271 (30): 18181–7. doi:10.1074/jbc.271.30.18181. PMID 8663458.
- Yoshida H, Haze K, Yanagi H, Yura T, Mori K (December 1998). "Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors". The Journal of Biological Chemistry. 273 (50): 33741–9. doi:10.1074/jbc.273.50.33741. PMID 9837962.
- Kandeil, Mohamed A.; Hashem, Reem M.; Mahmoud, Mohamed O.; Hetta, Mona H.; Tohamy, Mohamed A. (2019). "Zingiber officinale extract and omega-3 fatty acids ameliorate endoplasmic reticulum stress in a nonalcoholic fatty liver rat model". Journal of Food Biochemistry. 43 (12): e13076. doi:10.1111/jfbc.13076. hdl:2027.42/152724. ISSN 1745-4514. PMID 31608477. S2CID 204544806.
- Feyen, Dries A. M.; Perea-Gil, Isaac; Maas, Renee G. C.; Harakalova, Magdalena; Gavidia, Alexandra A.; Arthur Ataam, Jennifer; Wu, Ting-Hsuan; Vink, Aryan; Pei, Jiayi; Vadgama, Nirmal; Suurmeijer, Albert J. (2021-08-03). "Unfolded Protein Response as a Compensatory Mechanism and Potential Therapeutic Target in PLN R14del Cardiomyopathy". Circulation. 144 (5): 382–392. doi:10.1161/CIRCULATIONAHA.120.049844. ISSN 1524-4539. PMC 8667423. PMID 33928785.
- "Kitamura,M". Archived from the original on 2012-02-10. Retrieved 2008-02-06.
- Datan E, Roy SG, Germain G, Zali N, McLean JE, Golshan G, et al. (March 2016). "Dengue-induced autophagy, virus replication and protection from cell death require ER stress (PERK) pathway activation". Cell Death & Disease. 7 (e2127): e2127. doi:10.1038/cddis.2015.409. PMC 4823927. PMID 26938301.
- Roberson EC, Tully JE, Guala AS, Reiss JN, Godburn KE, Pociask DA, et al. (May 2012). "Influenza induces endoplasmic reticulum stress, caspase-12-dependent apoptosis, and c-Jun N-terminal kinase-mediated transforming growth factor-β release in lung epithelial cells". American Journal of Respiratory Cell and Molecular Biology. 46 (5): 573–81. doi:10.1165/rcmb.2010-0460OC. PMC 3359902. PMID 21799120.