Afterdepolarization
Afterdepolarizations are abnormal depolarizations of cardiac myocytes that interrupt phase 2, phase 3, or phase 4 of the cardiac action potential in the electrical conduction system of the heart. Afterdepolarizations may lead to cardiac arrhythmias. Afterdepolarization is commonly a consequence of myocardial infarction, cardiac hypertrophy, or heart failure.[1] It may also result from congenital mutations associated with calcium channels and sequestration.[2]
Early afterdepolarizations
Early afterdepolarizations (EADs) occur with abnormal depolarization during phase 2 or phase 3, and are caused by an increase in the frequency of abortive action potentials before normal repolarization is completed.[1] EADs most commonly originate in mid-myocardial cells and Purkinje fibers, but can develop in other cardiac cells that carry an action potential. Phase 2 may be interrupted due to augmented opening of calcium channels, while phase 3 interruptions are due to the opening of sodium channels. Early afterdepolarizations can result in torsades de pointes, tachycardia, and other arrhythmias.[3] EADs can be triggered by hypokalemia and drugs that prolong the QT interval, including class Ia and III antiarrhythmic agents, as well as catecholamines.[1]
Afterhyperpolarizations can also occur in cortical pyramidal neurons. There, they typically follow an action potential and are mediated by voltage gated sodium or chloride channels. This phenomenon requires potassium channels to close quickly to limit repolarization. It is responsible for the difference between regular spiking and intrinsically bursting pyramidal neurons.[4]
Delayed afterdepolarizations
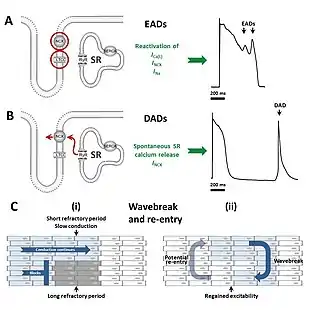
Delayed afterdepolarizations (DADs) begin during phase 4, after repolarization is completed but before another action potential would normally occur via the normal conduction systems of the heart. They are due to elevated cytosolic calcium concentrations, classically seen with digoxin toxicity.[5][6] The overload of the sarcoplasmic reticulum may cause spontaneous Ca2+ release after repolarization, causing the released Ca2+ to exit the cell through the 3Na+/Ca2+-exchanger. This results in a net depolarizing current. The classical feature is Bidirectional ventricular tachycardia. Also seen in catecholaminergic polymorphic ventricular tachycardia (CPVT). Delayed afterdepolarization is also seen in myocardial infarction. Purkinje fibers which survive myocardial infarction remain partially depolarized due to its high concentration of cations.[7] Partially depolarized tissue fires rapidly resulting in delayed after depolarization.[1]
References
- Antzelevitch, Charles; Burashnikov, Alexander (2011-03-01). "Overview of Basic Mechanisms of Cardiac Arrhythmia". Cardiac Electrophysiology Clinics. 3 (1): 23–45. doi:10.1016/j.ccep.2010.10.012. ISSN 1877-9182. PMC 3164530. PMID 21892379.
- Priori, S. G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G. A. (2001-01-16). "Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia". Circulation. 103 (2): 196–200. doi:10.1161/01.cir.103.2.196. ISSN 0009-7322. PMID 11208676.
- Cranefield, PF: The Conduction of the Cardiac Impulse. New York, Future Publishing Co. 1975
- Nelson Spruston, "Pyramidal Neurons: dendritic structure and synaptic integration", 2008. Nature Reviews. Neuroscience.
- Katzung, B: Basic and Clinical Pharmacology (10th ed.), chapter 14: "Agents Used in Cardiac Arrhythmias", The McGraw-Hill Companies, 2007, ISBN 978-0-07-145153-6
- Lilly, L: "Pathophysiology of Heart Disease", chapter 11: "Mechanisms of Cardiac Arrhthmias", Lippencott, Williams and Wilkens, 2007
- Lazzara, R.; el-Sherif, N.; Scherlag, B. J. (December 1973). "Electrophysiological properties of canine Purkinje cells in one-day-old myocardial infarction". Circulation Research. 33 (6): 722–734. doi:10.1161/01.res.33.6.722. ISSN 0009-7330. PMID 4762012.