Ectrodactyly–ectodermal dysplasia–cleft syndrome
Ectrodactyly–ectodermal dysplasia–cleft syndrome, or EEC, and also referred to as EEC syndrome[1] and split hand–split foot–ectodermal dysplasia–cleft syndrome[2]: 520 is a rare form of ectodermal dysplasia, an autosomal dominant disorder inherited as a genetic trait.[3]: 571 EEC is characterized by the triad of ectrodactyly, ectodermal dysplasia, and facial clefts.[4] Other features noted in association with EEC include vesicoureteral reflux, recurrent urinary tract infections,[5] obstruction of the nasolacrimal duct,[6] decreased pigmentation of the hair and skin, missing or abnormal teeth, enamel hypoplasia, absent punctae in the lower eyelids, photophobia, occasional cognitive impairment and kidney anomalies, and conductive hearing loss.[7][8]
| Ectrodactyly–ectodermal dysplasia–cleft syndrome | |
|---|---|
| Other names | EEC syndrome |
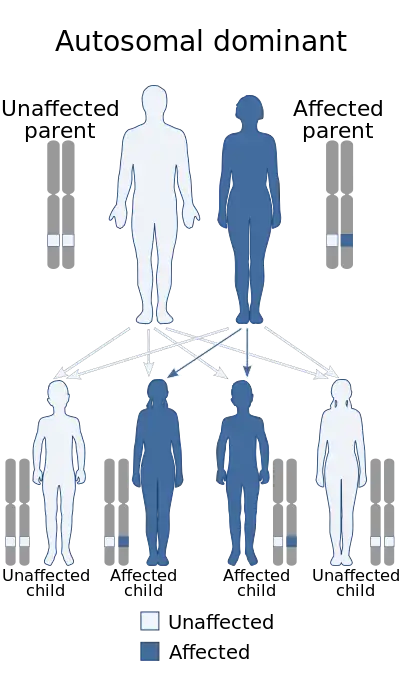 | |
| Ectrodactyly–ectodermal dysplasia–cleft syndrome is autosomal dominant | |
| Specialty | Medical genetics |
Presentation
Ectrodactyly
Ectrodactyly involves the deficiency or absence of one or more central digits of the hand or foot and is also known as split hand–split foot malformation (SHFM).[9] The hands and feet of people with ectrodactyly are often described as "claw-like" and may include only the thumb and one finger (usually either the little finger, ring finger, or a syndactyly of the two) with similar abnormalities of the feet.[6] It is sometimes referred to as "lobster claw" syndrome.
Ectodermal dysplasia describes abnormalities of structures derived from the embryonic ectoderm.[6] These abnormalities affect both the superficial ectodermal layer, as well as the mesectodermal layer constituted by the neural crest.[10]
Ectodermal dysplasia
Ectodermal dysplasia is characterized by absent sweat glands resulting in dry (hypohydrotic), often scale-like skin, sparse and usually coarse scalp hair that is often blonde, sparse eyebrows and eyelashes, and small brittle nails.[6] In addition, abnormalities of ectodermal derivatives, neuroectodermal derivatives, and mesectodermal derivatives are often found. The ectodermal derivative abnormalities can affect the epidermis including mammary, pituitary and sweat glands, as well as hairs, dental enamel, nails, lens, and the internal ear. Neuroectodermal derivatives that can be affected include sensory placodes, cutaneous pigmental cells, and hair buds. Mesectodermal derivatives affected can include the dermis, hypodermis, dentin, head muscles and conjunctival cells, cervicofacial vascular endothelial cells, and part of the maxillofacial skeleton.[10]
The hypohydrotic symptoms of ectodermal dysplasia described above are evidenced not only in the skin of affected individuals, but also in their phonation and voice production.[8] Because the vocal folds may not be as hydrated as is necessary during the adduction phase of vocal fold vibration (due to lack of lubrication), a complete seal may not be accomplished between the folds and mucosal wave movement may be disrupted.[11] This results in air escapement between the folds and the production of breathy voice, which often accompanies the skin abnormalities of ectodermal dysplasia.[6]
Facial clefting
There is much discrepancy in the literature regarding the exact nature of the facial clefting involved in EEC. Some authors claim that the clefting involved in EEC is always cleft lip +/- palate and use this marker as a means of distinguishing EEC from other syndromes, such as AEC syndrome (ankyloblepharon, ectodermal dysplasia, and clefting) in which other types of clefting are found.[7] Other authors include cleft palate only (CPO) in conjunction with ectrodactyly and ectodermal dysplasia as sufficient for a diagnosis of EEC.[8][12][13]
Speech deficits
The speech deficits associated with EEC syndrome are numerous. The clefting often causes hypernasal speech and velopharyngeal incompetence. Because of this, compensatory articulation strategies including retruded articulation and glottal compensation are often incorporated into the patient's speech. Articulation is further impaired by the numerous dental anomalies, including missing or malformed teeth found in EEC syndrome.[8]
Language deficits are also associated with EEC syndrome and are attributed to two factors. Conductive hearing loss due to ossicular anomalies is often encountered in patients with EEC syndrome, which can have significant impacts on language acquisition. Also, the impaired cognitive functioning that sometimes accompanies EEC can inhibit language acquisition.[8]
Embryology
The ectodermal dysplasia associated with EEC syndrome arises from abnormalities in the embryonic ectoderm, as described above.[6] Very early in embryonic development, the embryonic stem cells differentiate into three types of cells: the ectoderm, mesoderm, and endoderm. It is from these three types of cells that all body organs originate. In general terms, ectodermal cells generate the skin, spinal cord, and teeth (as well as the numerous derivatives mentioned above). Mesodermal cells generate blood vessels, muscle and bone, and endodermal cells generate the lungs, the digestive system and the urinary system.[14]
There are two layers of mesoderm; intraembryonic and extraembryonic. As the intraembryonic layer grows laterally, it becomes continuous with the extraembryonic layer, forming the chorion (contributing to the blood supply). At the same time during embryonic development, the ectoderm begins to thicken and fold upward, forming the neural folds, which eventually meet to form the neural tube and neural crest.[15] Because these two events occur at roughly the same time in embryological development, abnormalities found in this syndrome can involve not only the ectodermal cells, but also disruption to development in the mesectodermal layer constituted by the neural crest.[4][10]
"What these structures have in common is that their development and morphogenesis depends on the signaling between specialized ectodermal cells and the underlying mesoderm. Epithelial-mesenchymal interactions between the apical ectodermal ridge (AER) and the underlying mesenchyme, denoted the progress zone, are required for normal morphogenesis of the limb.[4]
Management
Research
Current research regarding EEC syndrome is focused on the genetic components contributing to the presented traits found in patients with EEC. A normal human karyotype includes 22 pairs of autosomal or non-sex chromosomes and one pair of sex chromosomes, constituting a total of 46 chromosomes. During reproduction, each parent contributes 23 chromosomes; 22 autosomal chromosomes and one sex chromosome.[16] As stated above, EEC syndrome is an autosomal dominant disorder.[4] This means that there is an abnormal gene on one of the autosomal (non-sex) chromosomes from either parent. Because the gene is dominant, only one parent must contribute the abnormal gene for the child to inherit the disease and the contributing parent will usually have the disease, due to the expression of the dominant gene in the parent.[16] Some characteristics of autosomal dominant inheritance patterns include a vertical transmission pattern, meaning that the disease phenotype is seen in generation after generation. Also, the recurrence risk is 50% and there are an equal number of affected males and females. Though we can calculate the chance of inheritance of the gene, the degree of expression cannot be calculated.[8]
Genetics
Genetics research relating to EEC has made great strides in recent years, but many findings are currently being debated in the literature. Chromosome 19, within the region of D19S894 and D19S416 has been postulated as the locus for the abnormalities found in EEC syndrome. This is supported by reports (though conflicting) regarding an association of cleft lip +/- palate on locus 19q, which suggests that EEC could be an allelic variant.[13]
More recently, the p63 gene has been targeted in numerous studies.[4][5][7][12] The p63 gene is a homologue of the tumor suppressor gene p53,[12] though this is not indicative that patients with EEC are more likely to develop tumors. p63 mutations have been implicated in other human malformation conditions as well, including AEC or Hay–Wells syndrome, limb–mammary syndrome, ADULT syndrome, and non-syndromic split hand–split foot malformation. When comparing the data for these syndromes, each syndrome has a distinct pattern and type of mutations, with extensive genotype–phenotype correlations. Brunner and colleagues found that most of the p63 mutations associated with EEC "involve amino acid substitutions in the DNA binding domain common to all known p63 isoforms". The findings of their study propose that the most frequently mutated arginine codons associated with EEC are 204, 227, 279, 280, and 304, with these five amino acid mutations accounting for 75% of all reported cases of EEC syndrome.[7] Other studies have had similar findings. One study found three of the five listed amino acid mutations in their subjects and noted that when 200 control chromosomes were tested, these three mutant alleles were not present.[12]
Mutations
The mutations found in EEC are missense mutations,[7] meaning that there is a single amino acid change in the protein, as opposed to premature termination of protein synthesis, known as a nonsense mutation.[4] The frameshift mutation introduces a premature stop codon that affected the α isotope, but does not affect the β and γ isotopes of p63. From this, it can be concluded that mutant p63α isotopes seem to play a major role in the pathogenesis of EEC syndrome.[4] It seems that p63α is the predominant p63 isotope in epithelial basal cell layers,[12] which are the cell type often associated with the anomalies found in patients with EEC syndrome.
Genetic expression
EEC can be both familial and sporadic, both cases relating back to abnormalities of the p63 gene. This means that in some cases, EEC expresses de novo in a child of unaffected parents (sporadic) due to spontaneous mutation, in addition to the existing autosomal dominant inherited form. There seems to be significant interfamilial and intrafamilial variability in expressivity, more noticeably between rather than within families. Because of this variability, it is possible that there is more than one genetic locus involved in the actual manifestation of the syndrome in any given person.[12] Other notably proposed sections of the involved chromosome include 3q27,[4][7][12] and more highly disputed areas, including 7q11.2–q21.3[12][13]
A study supports the hypothesis of the p63 gene as the locus for the mutations associated with EEC syndrome. The study is known as the p63 knockout mice study, in which the phenotypes of p63-deficient mice are described. The description of the mice is as follows:
P63-deficient mice lack all squamous epithelia and their derivatives, including hair, whiskers, teeth, as well as the mammary, lacrimal, and salivary glands. Particularly striking are severe limb truncations with forelimbs showing a complete absence of the phalanges and carpals, and variable defects of ulnae and radiae and hindlimbs that are lacking altogether…The p63 mutations act in a dominant fashion in humans, giving rise to a phenotype that resembles that of p63 knockout mice.[4]
This striking data offers convincing support for the p63 gene hypothesis. This study is also cited in the demonstration that the growth and patterning of the underlying mesenchyme is highly dependent on the apical ectodermal ridge of the limbs, as well as the maxillary and mandibular branchial ectoderm that are so prominently disturbed in these mice.[12] All of these findings are consistent with the clinical presentation of EEC in humans and may explain the association of limb malformation and clefting that are found in this syndrome.
In vitro model of EEC
Modeling EEC syndrome in vitro has been achieved by reprogramming EEC fibroblasts carrying mutations R304W and R204W into induced pluripotent stem cell (iPSC) lines. EEC-iPSC recapitulated defective epidermal and corneal fates. This model further identified PRIMA-1MET, a small compound that was identified as a compound targeting and reactivating p53 mutants based on a cell-based screening for rescuing the apoptotic activity of p53, as efficient to rescue R304W mutation defect.[17] Of interest, similar effect had been observed on keratinocytes derived from the same patients.[18] PRIMA-1MET could become an effective therapeutic tool for EEC patients.[19]
Further genetic research is necessary to identify and rule out other possible loci contributing to EEC syndrome, though it seems certain that disruption of the p63 gene is involved to some extent. In addition, genetic research with an emphasis on genetic syndrome differentiation should prove to be very useful in distinguishing between syndromes that present with very similar clinical findings. There is much debate in current literature regarding clinical markers for syndromic diagnoses. Genetic findings could have great implications in clinical diagnosis and treatment of not only EEC, but also many other related syndromes.
See also
References
- Buss PW, Hughes HE, Clarke A (September 1995). "Twenty-four cases of the EEC syndrome: clinical presentation and management". Journal of Medical Genetics. 32 (9): 716–723. doi:10.1136/jmg.32.9.716. PMC 1051673. PMID 8544192.
- Freedberg, et al. (2003). Fitzpatrick's Dermatology in General Medicine. (6th ed.). McGraw-Hill. ISBN 0-07-138076-0.
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 0-7216-2921-0.
- Celli J, Duijf P, Hamel BC, et al. (1999). "Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome" (PDF). Cell. 99 (2): 143–153. doi:10.1016/S0092-8674(00)81646-3. PMID 10535733. S2CID 18193864.
- Ramirez D, Lammer EJ (2004). "Lacrimoauriculodentodigital syndrome with cleft lip/palate and renal manifestations". Cleft Palate-Craniofacial Journal. 41 (5): 501–506. doi:10.1597/03-080.1. PMID 15352854. S2CID 20874635.
- Peterson-Falzone, Sally J.; Mary A. Hardin-Jones; Michael P. Karnell; Betty Jane McWilliams (2001). Cleft Palate Speech. Mosby. ISBN 978-0-8151-3153-3.
- Brunner HG, Hamel BC, Van Bokhoven H (2002). "The p63 gene in EEC and other syndromes". Journal of Medical Genetics. 39 (6): 377–381. doi:10.1136/jmg.39.6.377. PMC 1735150. PMID 12070241.
- Shprintzen, Robert J. (1997). Genetics, Syndromes, and Communication Disorders. Singular Pub. Group. ISBN 978-1-56593-620-1.
- Moerman P, Fryns JP (1996). "Ectodermal dysplasia, Rapp–Hodgkin type in a mother and severe ectrodactyly–ectodermal dysplasia–clefting syndrome (EEC) in her child". American Journal of Medical Genetics. 63 (3): 479–481. doi:10.1002/(SICI)1096-8628(19960614)63:3<479::AID-AJMG12>3.0.CO;2-J. PMID 8737656.
- Ruhin B, Martinot V, Lafforgue P, Catteau B, Manouvrier-Hanu S, Ferri J (2001). "Pure ectodermal dysplasia: retrospective study of 16 cases and literature review". Cleft Palate-Craniofacial Journal. 38 (5): 504–518. doi:10.1597/1545-1569(2001)038<0504:PEDRSO>2.0.CO;2. PMID 11522173.
- Peterson-Falzone SJ, Caldarelli DD, Landahl KL (1981). "Abnormal laryngeal vocal quality in ectodermal dysplasia". Archives of Otolaryngology. 107 (5): 300–304. doi:10.1001/archotol.1981.00790410038010. PMID 7224950.
- Barrow LL, van Bokhoven H, Daack-Hirsch S, et al. (2002). "Analysis of the p63 gene in classical EEC syndrome, related syndromes, and non-syndromic orofacial clefts". Journal of Medical Genetics. 39 (8): 559–566. doi:10.1136/jmg.39.8.559. PMC 1735218. PMID 12161593.
- O'Quinn JR, Hennekam RC, Jorde LB, Bamshad M (1998). "Syndromic ectrodactyly with severe limb, ectodermal, urogenital, and palatal defects maps to chromosome 19". American Journal of Human Genetics. 62 (1): 130–135. doi:10.1086/301687. PMC 1376811. PMID 9443880.
- Batshaw, Mark L. (2002). Children with Disabilities. Baltimore: Paul H. Brookes. ISBN 978-1-55766-581-2.
- Zemlin, Willard R. (1981). Speech and Hearing Science: Anatomy and Physiology. Prentice-Hall.
- Hall, Judith G. (August 2007). "Chromosomes and Genes". Merck. Retrieved 2008-10-01.
- Shalom Feuerstein R.; et al. (2012). "Impaired epithelial differentiation of induced pluripotent stem cells from EEC patients is rescued by APR-246/PRIMA-1MET". Proceedings of the National Academy of Sciences, USA. 110 (6): 2152–2156. doi:10.1073/pnas.1201753109. PMC 3568301. PMID 23355677.
- Shen J, van den Bogaard EH, Kouwenhoven EN, Bykov VJ, Rinne T, Zhang Q, Tjabringa GS, Gilissen C, van Heeringen SJ, Schalkwijk J, van Bokhoven H, Wiman KG, Zhou H (2013). "APR-246/PRIMA-1(MET) rescues epidermal differentiation in skin keratinocytes derived from EEC syndrome patients with p63 mutations". Proceedings of the National Academy of Sciences, USA. 110 (6): 2157–2162. Bibcode:2013PNAS..110.2157S. doi:10.1073/pnas.1201993110. PMC 3568378. PMID 23355676.
- Zhou H, Aberdam D (2013). "A step closer toward therapies for p63-related disorders". Rare Diseases. 1: e24247. doi:10.4161/rdis.24247. PMC 3932939. PMID 25002990.