Fumarylacetoacetate hydrolase
Fumarylacetoacetase is an enzyme that in humans is encoded by the FAH gene located on chromosome 15. The FAH gene is thought to be involved in the catabolism of the amino acid phenylalanine in humans.[5][6][7]
| FAH | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | FAH, Fumarylacetoacetase, fumarylacetoacetate hydrolase | ||||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | OMIM: 613871 MGI: 95482 HomoloGene: 110 GeneCards: FAH | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||




Function
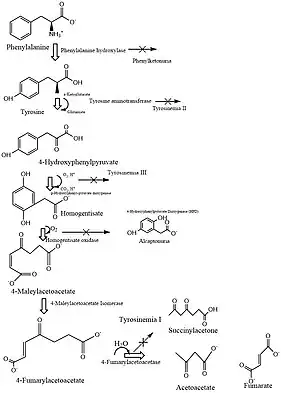
Fumarylacetoacetate hydrolase (FAH) is a protein homodimer which cleaves fumarylacetoacetate at its carbon-carbon bond during a hydrolysis reaction.[8] As a critical enzyme in phenylalanine and tyrosine metabolism, 4-Fumarylacetoacetate hydrolase catalyzes the final step in the catabolism of 4-fumarylacetoacetate and water into acetoacetate, fumarate, and H+ respectively.[9] These hydrolytic reactions are essential during aromatic amino acid human metabolism. Furthermore, FAH does not share known protein sequence homologs with other nucleotides or amino acids.[8]
Reaction mechanism
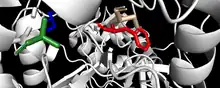
The active site of FAH contains Ca2+ which acts to bind the substrate and a Glu-His-Water catalytic triad functions where the imidaxole ring of His133 activates a nucleophilic water molecule to attack the carbon-carbon bond of fumarylactoacetate thus forming fumarate and acetoacetate.[11] Similar to Phenylalanine-associated pathways, the reaction molecular basis is critical in mammalian metabolism, as evidenced by the observed liver enzyme activity in FAH deficiency during hereditary tyrosinemia type 1.[12][13] In humans, this enzyme is mainly expressed in the liver. FAH is among the amino acid hydroxylases.[10] Tyrosine aminotransferase (TAT), 4-hydroxyphenylpyruvate dioxygenase (HPD), homogentisate 1,2-dioxygenase (HGD), phenylalanine-4-hydroxylase (PAH), maleylacetoacetate isomerase (GSTZ1),and other amino acid catabolic hydroxylases are coupled in the biological process of hydroxylases as well.[7][14] The FAH subpathway constitutes a part of the main amino-acid L-phenylalanine degradation.[10] For ingested phenylalanine, turnover for FAH protein synthesis is directly linked to treatment based methodology.
Mutations
The activity of human liver fumarylacetoacetate fumarylhydrolase has been determined with fumarylacetoacetate as the substrate.[15] As an inborn error of metabolism, Tyrosinemia type I stems from a deficiency in the enzymatic catabolic pathway of fumarylacetoacetate hydrolase (FAH). Currently, the mutations reported include silent mutations, amino acid replacements within single base substitutions, nonsense codons, and splicing defects.[15][16][17] Mutations spread across the FAH gene observes clusters of amino acid residues such as alanine and aspartic acid residues.[15] Hereditary tyrosinemia type 1 is a metabolic disorder with an autosomal recessive mode of inheritance. The disease is caused by a deficiency of fumarylacetoacetate hydrolase (FAH), the last enzyme in the degradation pathway of tyrosine. Hereditary tyrosinemia type 1 manifests in either an acute or a chronic form.[18][14] However, symptoms may appear in heterozygote mutations in the FAH gene as documented in case of a 12‐year‐old American boy with chronic tyrosinemia type 1.[19] Specifically, maternal alleles for codon 234 exhibit this mutation which changes a tryptophan to a glycine. This possibly suggests HT1 missense mutations also inhibiting enzymatic activity.[18] This is also attributed to observed clustering between amino acid residue active sites 230 and 250 among hundreds of other mutations in the FAH gene.[17] Currently, FAH gene correlation with HT1 does not associate clinical phenotype with genotype.[18][15][16]
Symptoms
Possible disease symptom is the development of Hereditary tyrosinemia type 1 (HT1).[15] Caused by the lack of fumarylacetoacetate hydrolase (FAH), the last enzyme of the tyrosine catabolic pathway, HT 1 is inherited as a rare autosomal recessive disease with a prevalence in Europe of 1 : 50000.[15][18] However, in isolated parts of Quebec's provinces, the frequency can be as high as 1 : 2000 with a carrier rate of 1:20 possibly due to a single founder mutation.[5][17] FAH deficiency leads to an accumulation of alkylafing metabolites that cause damage to the liver. The disorder presents as an acute, chronic or intermediate mild phenotype. The acute form manifests itself within the first half year and is characterized by liver failure, renal damage, and possibly death in the first year of life.[20] The chronic form has an age of onset of more than one year after birth;[21] rickets and progressive liver disease often lead to the development of hepatocellular carcinoma.[18] Other symptoms can include renal tubular injury, hepatic necrosis, episodic weakness, seizures. Renal Fanconi syndrome and Porphyric crises are also cited in addition to liver and renal damage.[18]
Treatment
Currently, there is no cure for tyrosinemia type 1. Diagnosed individuals need special dietary restriction all throughout life for the amino acids, phenylalanine and tyrosine.[22][23] Affected individuals may also be treated with a FDA-approved medication called nitisinone. Recommended treatment should begin as early as possible when the condition is diagnosed. Bacterial inhibition assay, such as the Guthrie Test, can screen newborns[21] for FAH deficiency in addition to increased phenylalanine levels.[23][24] Other diagnostic methods include measurements with tandem mass spectrometry fragmentation. Some individuals require a liver transplant if liver disease progresses into advanced development before dietary treatment begins.
Structure
A complete FAH genotype has been previously established.[10] All possible bands show two deleterious mutations. The effect of these mutations for the majority of the abnormalities on the FAH mRNA have been analysed. The identification of the gene defects on both alleles enables an initial genotype-phenotype analysis for chronic, subacute and acute HT 1 patients. The FAH gene is located on the chromosome 15q25.1 region and contains 14 exons. It encodes a protein that is 46kDa in height.[8] Multiple isoforms of the protein have been discovered that arose from alternative splicing. The gene is mainly expressed in the liver and the kidney.
References
- GRCh38: Ensembl release 89: ENSG00000103876 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000030630 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Phaneuf D, Labelle Y, Bérubé D, Arden K, Cavenee W, Gagné R, Tanguay RM (March 1991). "Cloning and expression of the cDNA encoding human fumarylacetoacetate hydrolase, the enzyme deficient in hereditary tyrosinemia: assignment of the gene to chromosome 15". American Journal of Human Genetics. 48 (3): 525–35. PMC 1682993. PMID 1998338.
- Agsteribbe E, van Faassen H, Hartog MV, Reversma T, Taanman JW, Pannekoek H, Evers RF, Welling GM, Berger R (April 1990). "Nucleotide sequence of cDNA encoding human fumarylacetoacetase". Nucleic Acids Research. 18 (7): 1887. doi:10.1093/nar/18.7.1887. PMC 330610. PMID 2336361.
- "Entrez Gene: FAH fumarylacetoacetate hydrolase (fumarylacetoacetase)".
- Timm DE, Mueller HA, Bhanumoorthy P, Harp JM, Bunick GJ (September 1999). "Crystal structure and mechanism of a carbon-carbon bond hydrolase". Structure. 7 (9): 1023–33. doi:10.1016/s0969-2126(99)80170-1. PMID 10508789.
- Universal protein resource accession number P16930 for "FAH - Fumarylacetoacetase - Homo sapiens (Human) - FAH gene & protein" at UniProt.
- Tanguay RM, Valet JP, Lescault A, Duband JL, Laberge C, Lettre F, Plante M (August 1990). "Different molecular basis for fumarylacetoacetate hydrolase deficiency in the two clinical forms of hereditary tyrosinemia (type I)". American Journal of Human Genetics. 47 (2): 308–16. PMC 1683717. PMID 2378356.
- Bateman RL, Bhanumoorthy P, Witte JF, McClard RW, Grompe M, Timm DE (May 2001). "Mechanistic inferences from the crystal structure of fumarylacetoacetate hydrolase with a bound phosphorus-based inhibitor". The Journal of Biological Chemistry. 276 (18): 15284–91. doi:10.1074/jbc.M007621200. PMID 11154690.
- Kvittingen, E.A.; Jellum, E.; Stokke, O. (1981-09-24). "Assay of fumarylacetoacetate fumarylhydrolase in human liver—deficient activity in a case of hereditary tyrosinemia". Clinica Chimica Acta. 115 (3): 311–319. doi:10.1016/0009-8981(81)90244-8. PMID 7296877.
- Kvittingen, E.A.; Jellum, E.; Stokke, O. (1981-09-24). "Assay of fumarylacetoacetate fumarylhydrolase in human liver—deficient activity in a case of hereditary tyrosinemia". Clinica Chimica Acta. 115 (3): 311–319. doi:10.1016/0009-8981(81)90244-8. ISSN 0009-8981. PMID 7296877.
- Sniderman King L, Trahms C, Scott CR (July 2006). "Tyrosinemia Type I". In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A (eds.). GeneReviews. Seattle: University of Washington. PMID 20301688.
- Labelle Y, Phaneuf D, Leclerc B, Tanguay RM (July 1993). "Characterization of the human fumarylacetoacetate hydrolase gene and identification of a missense mutation abolishing enzymatic activity". Human Molecular Genetics. 2 (7): 941–6. doi:10.1093/hmg/2.7.941. PMID 8364576.
- Rootwelt H, Berger R, Gray G, Kelly DA, Coşkun T, Kvittingen EA (October 1994). "Novel splice, missense, and nonsense mutations in the fumarylacetoacetase gene causing tyrosinemia type 1". American Journal of Human Genetics. 55 (4): 653–8. PMC 1918286. PMID 7942842.
- St-Louis, Maryse; Tanguay, Robert M. (1997). "Mutations in the fumarylacetoacetate hydrolase gene causing hereditary tyrosinemia type I: Overview". Human Mutation. 9 (4): 291–299. doi:10.1002/(sici)1098-1004(1997)9:4<291::aid-humu1>3.0.co;2-9. ISSN 1059-7794. PMID 9101289. S2CID 21881874.
- Ploos van Amstel JK, Bergman AJ, van Beurden EA, Roijers JF, Peelen T, van den Berg IE, Poll-The BT, Kvittingen EA, Berger R (January 1996). "Hereditary tyrosinemia type 1: novel missense, nonsense and splice consensus mutations in the human fumarylacetoacetate hydrolase gene; variability of the genotype-phenotype relationship". Human Genetics. 97 (1): 51–9. doi:10.1007/BF00218833. PMID 8557261. S2CID 20070794.
- Hahn SH, Krasnewich D, Brantly M, Kvittingen EA, Gahl WA (1995). "Heterozygosity for an exon 12 splicing mutation and a W234G missense mutation in an American child with chronic tyrosinemia type 1". Human Mutation. 6 (1): 66–73. doi:10.1002/humu.1380060113. PMID 7550234. S2CID 41460261.
- Phaneuf D, Lambert M, Laframboise R, Mitchell G, Lettre F, Tanguay RM (October 1992). "Type 1 hereditary tyrosinemia. Evidence for molecular heterogeneity and identification of a causal mutation in a French Canadian patient". The Journal of Clinical Investigation. 90 (4): 1185–92. doi:10.1172/JCI115979. PMC 443158. PMID 1401056.
- Mohamed S, Kambal MA, Al Jurayyan NA, Al-Nemri A, Babiker A, Hasanato R, Al-Jarallah AS (September 2013). "Tyrosinemia type 1: a rare and forgotten cause of reversible hypertrophic cardiomyopathy in infancy". BMC Research Notes. 6: 362. doi:10.1186/1756-0500-6-362. PMC 3846631. PMID 24016420.
- Online Mendelian Inheritance in Man (OMIM): Tyrosinemia, Type I; TYRSN1 - 276700
- Chinsky JM, Singh R, Ficicioglu C, van Karnebeek CD, Grompe M, Mitchell G, Waisbren SE, Gucsavas-Calikoglu M, Wasserstein MP, Coakley K, Scott CR (December 2017). "Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations". Genetics in Medicine. 19 (12): 1380–1395. doi:10.1038/gim.2017.101. PMC 5729346. PMID 28771246.
- "Tyrosinemia type 1 | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2018-12-13.
Further reading
- St-Louis M, Tanguay RM (1997). "Mutations in the fumarylacetoacetate hydrolase gene causing hereditary tyrosinemia type I: overview". Human Mutation. 9 (4): 291–9. doi:10.1002/(SICI)1098-1004(1997)9:4<291::AID-HUMU1>3.0.CO;2-9. PMID 9101289. S2CID 21881874.
- Phaneuf D, Lambert M, Laframboise R, Mitchell G, Lettre F, Tanguay RM (October 1992). "Type 1 hereditary tyrosinemia. Evidence for molecular heterogeneity and identification of a causal mutation in a French Canadian patient". The Journal of Clinical Investigation. 90 (4): 1185–92. doi:10.1172/JCI115979. PMC 443158. PMID 1401056.
- Tanguay RM, Valet JP, Lescault A, Duband JL, Laberge C, Lettre F, Plante M (August 1990). "Different molecular basis for fumarylacetoacetate hydrolase deficiency in the two clinical forms of hereditary tyrosinemia (type I)". American Journal of Human Genetics. 47 (2): 308–16. PMC 1683717. PMID 2378356.
- Laberge C, Grenier A, Valet JP, Morissette J (August 1990). "Fumarylacetoacetase measurement as a mass-screening procedure for hereditary tyrosinemia type I". American Journal of Human Genetics. 47 (2): 325–8. PMC 1683713. PMID 2378358.
- Kvittingen EA, Halvorsen S, Jellum E (July 1983). "Deficient fumarylacetoacetate fumarylhydrolase activity in lymphocytes and fibroblasts from patients with hereditary tyrosinemia". Pediatric Research. 17 (7): 541–4. doi:10.1203/00006450-198307000-00005. PMID 6622096.
- Kvittingen EA, Jellum E, Stokke O (September 1981). "Assay of fumarylacetoacetate fumarylhydrolase in human liver-deficient activity in a case of hereditary tyrosinemia". Clinica Chimica Acta; International Journal of Clinical Chemistry. 115 (3): 311–9. doi:10.1016/0009-8981(81)90244-8. PMID 7296877.
- Hahn SH, Krasnewich D, Brantly M, Kvittingen EA, Gahl WA (1995). "Heterozygosity for an exon 12 splicing mutation and a W234G missense mutation in an American child with chronic tyrosinemia type 1". Human Mutation. 6 (1): 66–73. doi:10.1002/humu.1380060113. PMID 7550234. S2CID 41460261.
- St-Louis M, Poudrier J, Phaneuf D, Leclerc B, Laframboise R, Tanguay RM (February 1995). "Two novel mutations involved in hereditary tyrosinemia type I". Human Molecular Genetics. 4 (2): 319–20. doi:10.1093/hmg/4.2.319. PMID 7757089.
- Kato S, Sekine S, Oh SW, Kim NS, Umezawa Y, Abe N, Yokoyama-Kobayashi M, Aoki T (December 1994). "Construction of a human full-length cDNA bank". Gene. 150 (2): 243–50. doi:10.1016/0378-1119(94)90433-2. PMID 7821789.
- Rootwelt H, Berger R, Gray G, Kelly DA, Coşkun T, Kvittingen EA (October 1994). "Novel splice, missense, and nonsense mutations in the fumarylacetoacetase gene causing tyrosinemia type 1". American Journal of Human Genetics. 55 (4): 653–8. PMC 1918286. PMID 7942842.
- Rootwelt H, Brodtkorb E, Kvittingen EA (December 1994). "Identification of a frequent pseudodeficiency mutation in the fumarylacetoacetase gene, with implications for diagnosis of tyrosinemia type I". American Journal of Human Genetics. 55 (6): 1122–7. PMC 1918441. PMID 7977370.
- Rootwelt H, Chou J, Gahl WA, Berger R, Coşkun T, Brodtkorb E, Kvittingen EA (June 1994). "Two missense mutations causing tyrosinemia type 1 with presence and absence of immunoreactive fumarylacetoacetase". Human Genetics. 93 (6): 615–9. doi:10.1007/BF00201558. PMID 8005583. S2CID 9382582.
- Grompe M, St-Louis M, Demers SI, al-Dhalimy M, Leclerc B, Tanguay RM (August 1994). "A single mutation of the fumarylacetoacetate hydrolase gene in French Canadians with hereditary tyrosinemia type I". The New England Journal of Medicine. 331 (6): 353–7. doi:10.1056/NEJM199408113310603. PMID 8028615.
- St-Louis M, Leclerc B, Laine J, Salo MK, Holmberg C, Tanguay RM (January 1994). "Identification of a stop mutation in five Finnish patients suffering from hereditary tyrosinemia type I". Human Molecular Genetics. 3 (1): 69–72. doi:10.1093/hmg/3.1.69. PMID 8162054.
- Grompe M, al-Dhalimy M (1993). "Mutations of the fumarylacetoacetate hydrolase gene in four patients with tyrosinemia, type I". Human Mutation. 2 (2): 85–93. doi:10.1002/humu.1380020205. PMID 8318997. S2CID 11639798.
- Labelle Y, Phaneuf D, Leclerc B, Tanguay RM (July 1993). "Characterization of the human fumarylacetoacetate hydrolase gene and identification of a missense mutation abolishing enzymatic activity". Human Molecular Genetics. 2 (7): 941–6. doi:10.1093/hmg/2.7.941. PMID 8364576.
- Labelle Y, Puymirat J, Tanguay RM (January 1993). "Localization of cells in the rat brain expressing fumarylacetoacetate hydrolase, the deficient enzyme in hereditary tyrosinemia type 1". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1180 (3): 250–6. doi:10.1016/0925-4439(93)90046-4. PMID 8422430.
- Ploos van Amstel JK, Bergman AJ, van Beurden EA, Roijers JF, Peelen T, van den Berg IE, Poll-The BT, Kvittingen EA, Berger R (January 1996). "Hereditary tyrosinemia type 1: novel missense, nonsense and splice consensus mutations in the human fumarylacetoacetate hydrolase gene; variability of the genotype-phenotype relationship". Human Genetics. 97 (1): 51–9. doi:10.1007/bf00218833. PMID 8557261. S2CID 20070794.