Farber disease
Farber disease (also known as Farber's lipogranulomatosis, acid ceramidase deficiency, "Lipogranulomatosis",[2] and ASAH1-related disorders) is an extremely rare, progressive, autosomal recessive lysosomal storage disease caused by a deficiency of the acid ceramidase enzyme. Acid ceramidase is responsible for breaking down ceramide into sphingosine and fatty acid.[3] When the enzyme is deficient, this leads to an accumulation of fatty material (called ceramide) in the lysosomes of the cells, leading to the signs and symptoms of this disorder.[4]
| Farber disease | |
|---|---|
| Other names | Acid ceramidase deficiency[1] |
| Specialty | Endocrinology |
Signs and symptoms
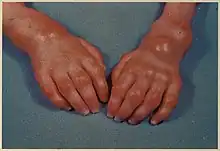
The symptoms of Farber disease develop over time. The onset of symptoms and how quickly they progress vary from person to person.[5]
The most common symptoms include:[4]
- Bumps under the skin located at pressure points and joints, also called subcutaneous nodules, lipogranulomas, or granulomas
- Swollen, painful joints with progressive limitation of range of motion resulting in contracture
- Hoarse voice/cry
Other symptoms observed in some individuals with Farber disease include:[5][3]
- Respiratory disease, e.g. lung infections, labored breathing, respiratory distress
- Central nervous system disease, e.g. developmental delay, muscle weakness, seizures
- Systemic inflammation
- Failure to thrive
- Bone disease, e.g. erosion of bone near joints, osteoporosis, peripheral osteolysis
- Enlarged liver (hepatomegaly)
- Eye disease, e.g. cherry-red spot, corneal opacities
Genetics

Farber disease is caused by variants in the ASAH1 gene. This gene codes for the acid ceramidase enzyme. Individuals with Farber disease have two copies of this gene that are not functioning properly leading to the enzyme deficiency. Over 73 different gene variants have been reported to cause Farber disease. No definitive genotype-phenotype correlations are known.[6]
Farber disease is inherited in an autosomal recessive manner. Affected individuals inherit one copy of the gene that is not functioning properly from each parent. Each parent is a called a carrier and has one copy of the gene that is functioning properly and one that is not. Siblings of individuals with Farber disease have a 25% chance to also have Farber disease, a 50% chance to be a carrier like the parents, and a 25% chance of being unaffected and not a carrier.[4]
The ASAH1 gene is also known to cause a condition called spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME).[4] Farber disease and SMA-PME have been classified as separate disorders, however more recent case reports have identified some individuals with overlapping symptoms of both disorders.[7][8] Some individuals with SMA symptoms without seizures have also been described.[9]
Diagnosis
Disease onset is typically in early infancy but may occur later in life. Children who have the classic form of Farber disease develop symptoms within the first few weeks to months of life.[4] Individuals with moderate or attenuated forms may develop symptoms at any time in childhood. Sometimes it is difficult to diagnose Farber disease because the symptoms can be misdiagnosed as Juvenile Idiopathic Arthritis (JIA).[10][11] Diagnosis is confirmed by molecular genetic testing of the ASAH1 gene or by measuring acid ceramidase enzyme activity.[4]
Treatment
There is no disease specific treatment for Farber disease. Anti-inflammatory medications, specifically tocilizumab (an interleukin-6 receptor inhibitor), has been shown to improve inflammation and pain in some patients.[12] Bone marrow transplant may improve granulomas (small masses of inflamed tissue) and inflammation in patients with little or no lung or nervous system complications.[13] Supportive therapies such as physical therapy, respiratory support, and mobility aids may be required.
Studies in cells and mice have shown proof-of-concept for enzyme replacement therapy for Farber disease.[14] Aceragen, a biopharmaceutical company, is currently developing an investigational enzyme replacement therapy with a clinical study planned for late 2022.
Prognosis
Children with the most severe forms of Farber disease generally die by age 2-3 years.[4] The life expectancy of individuals with moderate or attenuated forms is unknown. The oldest reported individuals living with Farber disease were in their 50s and 60s.[15]
Prevalence
To date, there have been approximately 200 reported cases of Farber disease and SMA-PME in the literature.[3] The disorders are ultra-rare and estimated to occur in fewer than one per million.[4]
Eponym
It is named for Sidney Farber.[16][17]
References
- RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Farber disease". www.orpha.net. Retrieved 17 April 2019.
- James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 978-0-7216-2921-6.
- Yu, FPS; Amintas, S; Levade, T; Medin, JA (20 July 2018). "Acid ceramidase deficiency: Farber disease and SMA-PME". Orphanet Journal of Rare Diseases. 13 (121): 20. doi:10.1186/s13023-018-0845-z. PMC 6053731. PMID 30029679.
- Dyment, DA; Bennett, SAL; Medin, JA; Levade, T; Adam, MP; Ardinger, HH; Pagon, RA; Wallace, SE; Bean, LJH; Mirzaa, G; Amemiya, A (2018). "ASAH1-Related Disorders". GeneReviews. PMID 29595935.
- "Farber disease". rarediseases.info.nih.gov. Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. Archived from the original on 2021-08-23. Retrieved 2021-08-17.
- Elsea, SH; Solyom, A; Martin, K; Harmatz, P; Mitchell, J; Lampe, C; Grant, C; Selim, L; Mungan, NO; Guelbert, N; Magnusson, B; Sundberg, E; Puri, R; Kapoor, S; Arslan, N; DiRocco, M; Zaki, M; Ozen, S; Mahmoud, IG; Ehlert, K; Hahn, A; Gokcay, G; Torcoletti, M; Ferreira, CR (September 2020). "ASAH1 pathogenic variants associated with acid ceramidase deficiency: Farber disease and spinal muscular atrophy with progressive myoclonic epilepsy". Human Mutation. 41 (9): 1469–1487. doi:10.1002/humu.24056. PMID 32449975. S2CID 218895424.
- Teoh, HL; Solyom, A; Schuchman, EH; Mowat, D; Roscioli, T; Farrar, M; Sampaio, H (October 2016). "Polyarticular Arthritis and Spinal Muscular Atrophy in Acid Ceramidase Deficiency". Pediatrics. 138 (4): e20161068. doi:10.1542/peds.2016-1068. PMID 27650050. S2CID 2152446.
- Lee, BH; Mongiovi, P; Levade, T; Marston, B; Mountain, J; Ciafaloni, E (October 2020). "Spinal muscular atrophy and Farber disease due to ASAH1 variants: A case report". American Journal of Medical Genetics. Part A. 182 (10): 2369–2371. doi:10.1002/ajmg.a.61764. PMID 32627310. S2CID 220369745.
- Filosto, M; Aureli, M; Castellotti, B; Rinaldi, F; Schiumarini, D; Valsecchi, M; Lualdi, S; Mazzotti, R; Pensato, V; Rota, S; Gellera, C; Filocamo, M; Padovani, A (November 2016). "ASAH1 variant causing a mild SMA phenotype with no myoclonic epilepsy: a clinical, biochemical and molecular study". European Journal of Human Genetics. 24 (11): 1578–1583. doi:10.1038/ejhg.2016.28. PMC 5110045. PMID 27026573.
- Schuchman, Edward H.; Mitchell, John; Solyom, Alex (2 September 2017). "Morbidity and mortality associated with Farber disease and prospects for therapy". Expert Opinion on Orphan Drugs. 5 (9): 717–726. doi:10.1080/21678707.2017.1359086. S2CID 79980666.
- Moghadam, SH; Tavasoli, AR; Modaresi, M; Ziaee, V (1 December 2019). "Farber disease: report of three cases with joint involvement mimicking juvenile idiopathic arthritis". Journal of Musculoskeletal & Neuronal Interactions. 19 (4): 521–525. PMC 6944811. PMID 31789304.
- Mitchell, John; Solyom, Alexander; Makay, Balahan; Arslan, Nur; Batu, Ezgi Deniz; Ozen, Seza; Hügle, Boris; Schuchman, Edward; Magnusson, Bo (February 2016). "Farber disease: Implications of anti-inflammatory treatment". Molecular Genetics and Metabolism. 117 (2): S81–S82. doi:10.1016/j.ymgme.2015.12.364.
- Ehlert, K; Levade, T; Di Rocco, M; Lanino, E; Albert, MH; Führer, M; Jarisch, A; Güngör, T; Ayuk, F; Vormoor, J (March 2019). "Allogeneic hematopoietic cell transplantation in Farber disease". Journal of Inherited Metabolic Disease. 42 (2): 286–294. doi:10.1002/jimd.12043. PMID 30815900. S2CID 73478264.
- He, X; Dworski, S; Zhu, C; DeAngelis, V; Solyom, A; Medin, JA; Simonaro, CM; Schuchman, EH (June 2017). "Enzyme replacement therapy for Farber disease: Proof-of-concept studies in cells and mice". BBA Clinical. 7: 85–96. doi:10.1016/j.bbacli.2017.02.001. PMC 5338723. PMID 28275553.
- Bonafé, L; Kariminejad, A; Li, J; Royer-Bertrand, B; Garcia, V; Mahdavi, S; Bozorgmehr, B; Lachman, RL; Mittaz-Crettol, L; Campos-Xavier, B; Nampoothiri, S; Unger, S; Rivolta, C; Levade, T; Superti-Furga, A (September 2016). "Brief Report: Peripheral Osteolysis in Adults Linked to ASAH1 (Acid Ceramidase) Mutations: A New Presentation of Farber's Disease". Arthritis & Rheumatology. 68 (9): 2323–7. doi:10.1002/art.39659. PMID 26945816. S2CID 37749661.
- synd/453 at Who Named It?
- Farber S (1952). "A lipid metabolic disorder: disseminated lipogranulomatosis; a syndrome with similarity to, and important difference from, Niemann-Pick and Hand-Schüller-Christian disease". American Journal of Diseases of Children. 84 (4): 499–500. PMID 12975849.