Active site
[1] In biology and biochemistry, the active site is the region of an enzyme where substrate molecules bind and undergo a chemical reaction. The active site consists of amino acid residues that form temporary bonds with the substrate, the binding site, and residues that catalyse a reaction of that substrate, the catalytic site. Although the active site occupies only ~10–20% of the volume of an enzyme,[2]: 19 it is the most important part as it directly catalyzes the chemical reaction. It usually consists of three to four amino acids, while other amino acids within the protein are required to maintain the tertiary structure of the enzymes.[3]
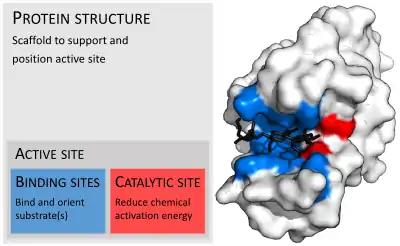
Each active site is evolved to be optimised to bind a particular substrate and catalyse a particular reaction, resulting in high specificity. This specificity is determined by the arrangement of amino acids within the active site and the structure of the substrates. Sometimes enzymes also need to bind with some cofactors to fulfil their function. The active site is usually a groove or pocket of the enzyme which can be located in a deep tunnel within the enzyme,[4] or between the interfaces of multimeric enzymes. An active site can catalyse a reaction repeatedly as residues are not altered at the end of the reaction (they may change during the reaction, but are regenerated by the end).[5] This process is achieved by lowering the activation energy of the reaction, so more substrates have enough energy to undergo reaction.
Binding site
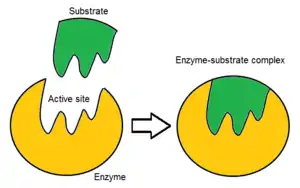
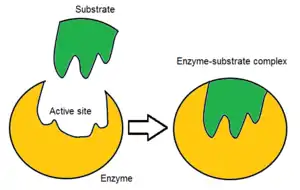
Usually, an enzyme molecule has only one active site, and the active site fits with one specific type of substrate. An active site contains a binding site that binds the substrate and orients it for catalysis. The orientation of the substrate and the close proximity between it and the active site is so important that in some cases the enzyme can still function properly even though all other parts are mutated and lose function.[6]
Initially, the interaction between the active site and the substrate is non-covalent and transient. There are four important types of interaction that hold the substrate in a defined orientation and form an enzyme-substrate complex (ES complex): hydrogen bonds, van der Waals interactions, hydrophobic interactions and electrostatic force interactions.[7]: 148 The charge distribution on the substrate and active site must be complementary, which means all positive and negative charges must be cancelled out. Otherwise, there will be a repulsive force pushing them apart. The active site usually contains non-polar amino acids, although sometimes polar amino acids may also occur.[3] The binding of substrate to the binding site requires at least three contact points in order to achieve stereo-, regio-, and enantioselectivity. For example, alcohol dehydrogenase which catalyses the transfer of a hydride ion from ethanol to NAD+ interacts with the substrate methyl group, hydroxyl group and the pro-(R) hydrogen that will be abstracted during the reaction.[7]: 149
In order to exert their function, enzymes need to assume their correct protein fold (native fold) and tertiary structure. To maintain this defined three-dimensional structure, proteins rely on various types of interactions between their amino acid residues. If these interactions are interfered with, for example by extreme pH values, high temperature or high ion concentrations, this will cause the enzyme to denature and lose its catalytic activity.
A tighter fit between an active site and the substrate molecule is believed to increase the efficiency of a reaction. If the tightness between the active site of DNA polymerase and its substrate is increased, the fidelity, which means the correct rate of DNA replication will also increase.[8] Most enzymes have deeply buried active sites, which can be accessed by a substrate via access channels.[4]
There are three proposed models of how enzymes fit their specific substrate: the lock and key model, the induced fit model, and the conformational selection model. The latter two are not mutually exclusive: conformational selection can be followed by a change in the enzyme's shape. Additionally, a protein may not wholly follow either model. Amino acids at the binding site of ubiquitin generally follow the induced fit model, whereas the rest of the protein generally adheres to conformational selection. Factors such as temperature likely influences the pathway taken during binding, with higher temperatures predicted to increase the importance of conformational selection and decrease that of induced fit.[9]
Lock and key hypothesis
This concept was suggested by the 19th-century chemist Emil Fischer. He proposed that the active site and substrate are two stable structures that fit perfectly without any further modification, just like a key fits into a lock. If one substrate perfectly binds to its active site, the interactions between them will be strongest, resulting in high catalytic efficiency.
As time went by, limitations of this model started to appear. For example, the competitive enzyme inhibitor methylglucoside can bind tightly to the active site of 4-alpha-glucanotransferase and perfectly fits into it. However, 4-alpha-glucanotransferase is not active on methylglucoside and no glycosyl transfer occurs. The Lock and Key hypothesis cannot explain this, as it would predict a high efficiency of methylglucoside glycosyl transfer due to its tight binding. Apart from competitive inhibition, this theory cannot explain the mechanism of action of non-competitive inhibitors either, as they do not bind to the active site but nevertheless influence catalytic activity.[10]
Induced fit hypothesis
Daniel Koshland's theory of enzyme-substrate binding is that the active site and the binding portion of the substrate are not exactly complementary.[11] The induced fit model is a development of the lock-and-key model and assumes that an active site is flexible and changes shape until the substrate is completely bound. This model is similar to a person wearing a glove: the glove changes shape to fit the hand. The enzyme initially has a conformation that attracts its substrate. Enzyme surface is flexible and only the correct catalyst can induce interaction leading to catalysis. Conformational changes may then occur as the substrate is bound. After the reaction products will move away from the enzyme and the active site returns to its initial shape. This hypothesis is supported by the observation that the entire protein domain could move several nanometers during catalysis. This movement of protein surface can create microenvironments that favour the catalysis.[6]
Conformational selection hypothesis
This model suggests that enzymes exist in a variety of conformations, only some of which are capable of binding to a substrate. When a substrate is bound to the protein, the equilibrium in the conformational ensemble shifts towards those able to bind ligands (as enzymes with bound substrates are removed from the equilibrium between the free conformations).[12]
Types of non-covalent interactions
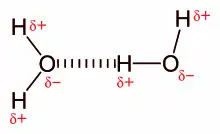
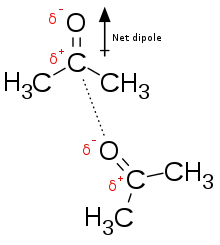
Electrostatic interaction: In an aqueous environment, the oppositely charged groups in amino acid side chains within the active site and substrates attract each other, which is termed electrostatic interaction. For example, when a carboxylic acid (R-COOH) dissociates into RCOO− and H+ ions, COO− will attract positively charged groups such as protonated guanidine side chain of arginine.
Hydrogen bond: A hydrogen bond is a specific type of dipole-dipole interaction between a partially positive hydrogen atom and a partially negative electron donor that contain a pair of electrons such as oxygen, fluorine and nitrogen. The strength of hydrogen bond depends on the chemical nature and geometric arrangement of each group.
Van der Waals force: Van der Waals force is formed between oppositely charged groups due to transient uneven electron distribution in each group. If all electrons are concentrated at one pole of the group this end will be negative, while the other end will be positive. Although the individual force is weak, as the total number of interactions between the active site and substrate is massive the sum of them will be significant.
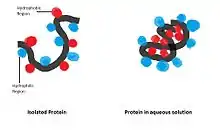
Hydrophobic interaction: Non-polar hydrophobic groups tend to aggregate together in the aqueous environment and try to leave from polar solvent. These hydrophobic groups usually have long carbon chain and do not react with water molecules. When dissolving in water a protein molecule will curl up into a ball-like shape, leaving hydrophilic groups in outside while hydrophobic groups are deeply buried within the centre.
Catalytic site
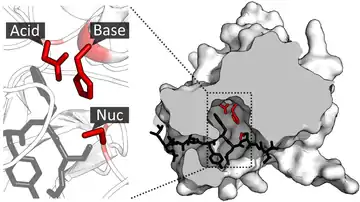
Once the substrate is bound and oriented to the active site, catalysis can begin. The residues of the catalytic site are typically very close to the binding site, and some residues can have dual-roles in both binding and catalysis.
Catalytic residues of the site interact with the substrate to lower the activation energy of a reaction and thereby make it proceed faster. They do this by a number of different mechanisms including the approximation of the reactants, nucleophilic/electrophilic catalysis and acid/base catalysis. These mechanisms will be explained below.
Mechanisms involved in Catalytic process
Approximation of the reactant
During enzyme catalytic reaction, the substrate and active site are brought together in a close proximity. This approach has various purposes. Firstly, when substrates bind within the active site the effective concentration of it significantly increases than in solution. This means the number of substrate molecules involved in the reaction is also increased. This process also reduces the desolvation energy required for the reaction to occur. In solution substrate molecules are surrounded by solvent molecules and energy is required for enzyme molecules to replace them and contact with the substrate. Since bulk molecules can be excluded from the active site this energy output can be minimised. Next, the active site is designed to reorient the substrate to reduce the activation energy for the reaction to occur. The alignment of the substrate, after binding, is locked in a high energy state and can proceed to the next step. In addition, this binding is favoured by entropy as the energy cost associated with solution reaction is largely eliminated since solvent cannot enter active site. In the end, the active site may manipulate the Molecular orbital of the substrate into a suitable orientation to reduce activation energy.[7]: 155–8
The electrostatic states of substrate and active site must be complementary to each other. A polarized negatively charged amino acid side chain will repel uncharged substrate. But if the transition state involves the formation of an ion centre then the side chain will now produce a favourable interaction.
Covalent catalysis
Many enzymes including serine protease, cysteine protease, protein kinase and phosphatase evolved to form transient covalent bonds between them and their substrates to lower the activation energy and allow the reaction to occur. This process can be divided into 2 steps: formation and breakdown. The former step is rate-limit step while the later step is needed to regenerate intact enzyme.[7]: 158
Nucleophilic catalysis: This process involves the donation of electrons from the enzyme's nucleophile to a substrate to form a covalent bond between them during the transition state. The strength of this interaction depends on two aspects.: the ability of the nucleophilic group to donate electrons and the electrophile to accept them. The former one is mainly affected by the basicity(the ability to donate electron pairs) of the species while the later one is in regard to its pKa. Both groups are also affected by their chemical properties such as polarizability, electronegativity and ionization potential. Amino acids that can form nucleophile including serine, cysteine, aspartate and glutamine.
Electrophilic catalysis: The mechanism behind this process is exactly same as nucleophilic catalysis except that now amino acids in active site act as electrophile while substrates are nucleophiles. This reaction usually requires cofactors as the amino acid side chains are not strong enough in attracting electrons.
Metal ions
Metal ions have multiple roles during the reaction. Firstly it can bind to negatively charged substrate groups so they will not repel electron pairs from active site's nucleophilic groups. It can attract negatively charged electrons to increase electrophilicity. It can also bridge between active site and substrate. At last, they may change the conformational structure of the substrate to favour reaction. [7]: 158
Acid/base catalysis
In some reactions, protons and hydroxide may directly act as acid and base in term of specific acid and specific base catalysis. But more often groups in substrate and active site act as Brønsted–Lowry acid and base. This is called general acid and general base theory. The easiest way to distinguish between them is to check whether the reaction rate is determined by the concentrations of the general acid and base. If the answer is yes then the reaction is the general type. Since most enzymes have an optimum pH of 6 to 7, the amino acids in the side chain usually have a pKa of 4~10. Candidate include aspartate, glutamate, histidine, cysteine. These acids and bases can stabilise the nucleophile or electrophile formed during the catalysis by providing positive and negative charges.[7]: 164–70
Conformational distortion
Quantitative studies of enzymatic reactions often found that the acceleration of chemical reaction speed cannot be fully explained by existing theories like the approximation, acid/base catalysis and electrophile/nucleophile catalysis. And there is an obvious paradox: in reversible enzymatic reaction if the active site perfectly fits the substrates then the backward reaction will be slowed since products cannot fit perfectly into the active site. So conformational distortion was introduced and argues that both active site and substrate can undergo conformational changes to fit with each other all the time.[7]: 170–5
Preorganised active site complementarity to the transition state
This theory is a little similar to the Lock and Key Theory, but at this time the active site is preprogrammed to bind perfectly to substrate in transition state rather than in ground state. The formation of transition state within the solution requires a large amount of energy to relocate solvent molecules and the reaction is slowed. So the active site can substitute solvent molecules and surround the substrates to minimize the counterproductive effect imposed by the solution. The presence of charged groups with the active site will attract substrates and ensure electrostatic complementarity.[7]: 176–8
Examples of enzyme catalysis mechanisms
In reality, most enzyme mechanisms involve a combination of several different types of catalysis.
Glutathione reductase
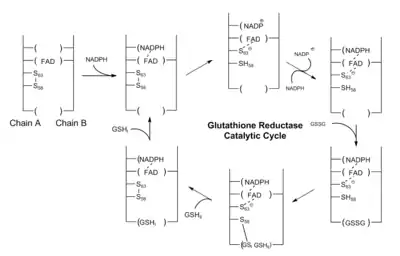
The role of glutathione(GSH) is to remove accumulated reactive oxygen species which may damage cells. During this process, its thiol side chain is oxidised and two glutathione molecules are connected by a disulphide bond to form a dimer(GSSG). In order to regenerate glutathione the disulphide bond has to be broken, In human cells, this is done by glutathione reductase(GR).
Glutathione reductase is a dimer that contains two identical subunits. It requires one NADP and one FAD as the cofactors. The active site is located in the linkage between two subunits. The NADPH is involved in the generation of FADH-. In the active site, there are two cysteine residues besides the FAD cofactor and are used to break the disulphide bond during the catalytic reaction. NADPH is bound by three positively charged residues: Arg-218, His-219 and Arg-224.
The catalytic process starts when the FAD is reduced by NADPH to accept one electron and from FADH−. It then attacks the disulphide bond formed between 2 cysteine residues, forming one SH bond and a single S− group. This S− group will act as a nucleophile to attack the disulphide bond in the oxidised glutathione(GSSG), breaking it and forming a cysteine-SG complex. The first SG− anion is released and then receives one proton from adjacent SH group and from the first glutathione monomer. Next the adjacent S− group attack disulphide bond in cysteine-SG complex and release the second SG− anion. It receives one proton in solution and forms the second glutathione monomer.
[2]: 137–9
Chymotrypsin
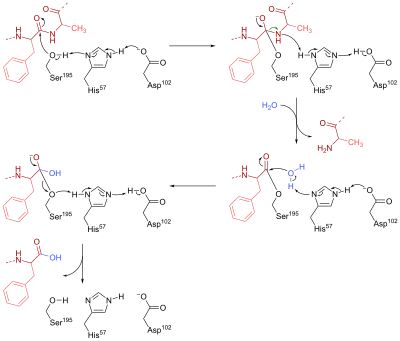
Chymotrypsin is a serine endopeptidase that is present in pancreatic juice and helps the hydrolysis of proteins and peptide.[2]: 84–6 It catalyzes the hydrolysis of peptide bonds in L-isomers of tyrosine, phenylalanine, and tryptophan. In the active site of this enzyme, three amino acid residues work together to form a catalytic triad which makes up the catalytic site. In chymotrypsin, these residues are Ser-195, His-57 and Asp-102.
The mechanism of chymotrypsin can be divided into two phases. First, Ser-195 nucleophilically attacks the peptide bond carbon in the substrate to form a tetrahedral intermediate. The nucleophilicity of Ser-195 is enhanced by His-57, which abstracts a proton from Ser-195 and is in turn stabilised by the negatively charged carboxylate group (RCOO−) in Asp-102. Furthermore, the tetrahedral oxyanion intermediate generated in this step is stabilised by hydrogen bonds from Ser-195 and Gly-193.
In the second stage, the R'NH group is protonated by His-57 to form R'NH2 and leaves the intermediate, leaving behind the acylated Ser-195. His-57 then acts as a base again to abstract one proton from a water molecule. The resulting hydroxide anion nucleophilically attacks the acyl-enzyme complex to form a second tetrahedral oxyanion intermediate, which is once again stabilised by H bonds. In the end, Ser-195 leaves the tetrahedral intermediate, breaking the CO bond that connected the enzyme to the peptide substrate. A proton is transferred to Ser-195 through His-57, so that all three amino acid return to their initial state.
Unbinding
Substrate unbinding is influenced by various factors. Larger ligands generally stay in the active site longer,[13] as do those with more rotatable bonds (although this may be a side effect of size).[14] When the solvent is excluded from the active site, less flexible proteins result in longer residence times. More hydrogen bonds shielded from the solvent also decrease unbinding.[13]
Cofactors

Enzymes can use cofactors as ‘helper molecules’. Coenzymes are referred to those non-protein molecules that bind with enzymes to help them fulfill their jobs. Mostly they are connected to the active site by non-covalent bonds such as hydrogen bond or hydrophobic interaction. But sometimes a covalent bond can also form between them. For example, the heme in cytochrome C is bound to the protein through thioester bond. In some occasions, coenzymes can leave enzymes after the reaction is finished. Otherwise, they permanently bind to the enzyme.[7]: 69 Coenzyme is a broad concept which includes metal ions, various vitamins and ATP. If an enzyme needs coenzyme to work itself, it is called an apoenzyme. In fact, it alone cannot catalyze reactions properly. Only when its cofactor comes in and binds to the active site to form holoenzyme does it work properly.
One example of the coenzyme is Flavin. It contains a distinct conjugated isoalloxazine ring system. Flavin has multiple redox states and can be used in processes that involve the transfer of one or two electrons. It can act as an electron acceptor in reaction, like the oxidation of NAD to NADH, to accept two electrons and form 1,5-dihydroflavin. On the other hand, it can form semiquinone(free radical) by accepting one electron, and then converts to fully reduced form by the addition of an extra electron. This property allows it to be used in one electron oxidation process.
Inhibitors
Inhibitors disrupt the interaction between enzyme and substrate, slowing down the rate of a reaction. There are different types of inhibitor, including both reversible and irreversible forms.
Competitive inhibitors are inhibitors that only target free enzyme molecules. They compete with substrates for free enzyme acceptor and can be overcome by increasing the substrate concentration. They have two mechanisms. Competitive inhibitors usually have structural similarities to the substrates and or ES complex. As a result, they can fit into the active site and trigger favourable interactions to fill in the space and block substrates from entry. They can also induce transient conformational changes in the active site so substrates cannot fit perfectly with it. After a short period of time, competitive inhibitors will drop off and leave the enzyme intact.
Inhibitors are classified as non-competitive inhibitors when they bind both free enzyme and ES complex. Since they do not compete with substrates for the active site, they cannot be overcome by simply increasing the substrate concentration. They usually bind to a different site on the enzyme and alter the 3-dimensional structure of the active site to block substrates from entry or leaving the enzyme.
Irreversible inhibitors are similar to competitive inhibitors as they both bind to the active site. However, irreversible inhibitors form irreversible covalent bonds with the amino acid residues in the active site and never leave. Therefore, the active site is occupied and the substrate cannot enter. Occasionally the inhibitor will leave but the catalytic site is permanently altered in shape. These inhibitors usually contain electrophilic groups like halogen substitutes and epoxides. As time goes by more and more enzymes are bound by irreversible inhibitors and cannot function anymore.
| Example | Binds active site? | Reduces rate of reaction? | |
|---|---|---|---|
| Competitive reversible inhibitor | HIV protease inhibitors | Yes | Yes |
| Non-competitive reversible inhibitor | Heavy metals such as lead and mercury | No | Yes |
| Irreversible inhibitor | Cyanide | Yes | Yes |
Examples of competitive and irreversible enzyme inhibitors
Competitive inhibitor: HIV protease inhibitor
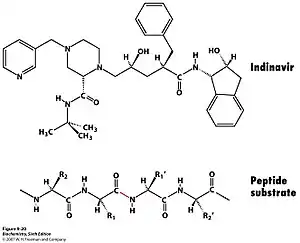
HIV protease inhibitors are used to treat patients having AIDS virus by preventing its DNA replication. HIV protease is used by the virus to cleave Gag-Pol polyprotein into 3 smaller proteins that are responsible for virion assembly, package and maturation. This enzyme targets the specific phenylalanine-proline cleave site within the target protein.[15] If HIV protease is switched off the virion particle will lose function and cannot infect patients. Since it is essential in viral replication and is absent in healthy human, it is an ideal target for drug development.
HIV protease belongs to aspartic protease family and has a similar mechanism. Firstly the aspartate residue activates a water molecule and turns it into a nucleophile. Then it attacks the carbonyl group within the peptide bond (NH-CO) to form a tetrahedral intermediate. The nitrogen atom within the intermediate receives a proton, forming an amide group and subsequent rearrangement leads to the breakdown of the bond between it and the intermediate and forms two products.[16]
Inhibitors usually contain a nonhydrolyzable hydroxyethylene or hydroxyethylamine groups that mimic the tetrahedral intermediate. Since they share a similar structure and electrostatic arrangement to the transition state of substrates they can still fit into the active site but cannot be broken down, so hydrolysis cannot occur.
Non-competitive inhibitor: Strychnine
Strychnine is a neurotoxin that causes death by affecting nerves that control muscular contraction and cause respiration difficulty. The impulse is transmitted between the synapse through a neurotransmitter called acetylcholine. It is released into the synapse between nerve cells and binds to receptors in the postsynaptic cell. Then an action potential is generated and transmitted through the postsynaptic cell to start a new cycle.
Glycine can inhibit the activity of neurotransmitter receptors, thus a larger amount of acetylcholinesterase is required to trigger an action potential. This makes sure that the generation of nerve impulses is tightly controlled. However, this control is broken down when strychnine is added. It inhibits glycine receptors(a chloride channel) and a much lower level of neurotransmitter concentration can trigger an action potential. Nerves now constantly transmit signals and cause excessive muscular contraction, leading to asphyxiation and death.[17]
Irreversible inhibitor: Diisopropyl fluorophosphate

Diisopropyl fluorophosphate (DIFP) is an irreversible inhibitor that blocks the action of serine protease. When it binds to the enzyme a nucleophilic substitution reaction occurs and releases one hydrogen fluoride molecule. The OH group in the active site acts as a nucleophile to attack the phosphorus in DIFP and form a tetrahedral intermediate and release a proton. Then the P-F bond is broken, one electron is transferred to the F atom and it leaves the intermediate as F− anion. It combines with a proton in solution to form one HF molecule. A covalent bond formed between the active site and DIFP, so the serine side chain is no longer available to the substrate.[18]
In drug discovery
Identification of active sites is crucial in the process of drug discovery. The 3-D structure of the enzyme is analysed to identify active site residues and design drugs which can fit into them. Proteolytic enzymes are targets for some drugs, such as protease inhibitors, which include drugs against AIDS and hypertension.[19] These protease inhibitors bind to an enzyme's active site and block interaction with natural substrates.[20] An important factor in drug design is the strength of binding between the active site and an enzyme inhibitor.[21] If the enzyme found in bacteria is significantly different from the human enzyme then an inhibitor can be designed against that particular bacterium without harming the human enzyme. If one kind of enzyme is only present in one kind of organism, its inhibitor can be used to specifically wipe them out.
Active sites can be mapped to aid the design of new drugs such as enzyme inhibitors. This involves the description of the size of an active site and the number and properties of sub-sites, such as details of the binding interaction.[19] Modern database technology called CPASS (Comparison of Protein Active Site Structures) however allows the comparison of active sites in more detail and the finding of structural similarity using software.[22]
Application of enzyme inhibitors
| Example | Mechanism of action | |
|---|---|---|
| Anti-bacterial agent | Penicillin | The bacterial cell wall is composed of peptidoglycan. During bacterial growth the present crosslinking of peptidoglycan fibre is broken, so new cell wall monomer can be integrated into the cell wall. Penicillin works by inhibiting the transpeptidase which is essential for the formation of crosslinks, so the cell wall is weakened and will burst open due to turgor pressure. |
| Anti-fungi agent | Azole | Ergosterol is a sterol that forms the cell surface membrane of the fungi. Azole can inhibit its biosynthesis by inhibiting the Lanosterol 14 alpha-demethylase, so no new ergosterol is produced and harmful 14α-lanosterol is accumulated within the cell. Also, azole may generate reactive oxygen species. |
| Anti-viral agent | Saquinavir | HIV protease is needed to cleave Gag-Pol polyprotein into 3 individual proteins so they can function properly and start viral packaging process. HIV protease inhibitors like Saquinavir inhibit it so no new mature viral particle can be made. |
| Insecticides | Physostigmine | In the animal nervous system, Acetylcholinesterase is required to break down the neurotransmitter acetylcholine into acetate and choline. Physostigmine binds to its active site and inhibits it, so impulse signal cannot be transmitted through nerves. This results in the death of insects as they lose control of muscle and heart function. |
| Herbicides | Cyclohexanedione | Cyclohexanedione targets the Acetyl-CoA carboxylase which is involved in the first step of fat synthesis: ATP-dependent carboxylation of acetyl-CoA to malonyl-CoA. Lipids are important in making up the cell membrane. |
Allosteric sites
An allosteric site is a site on an enzyme, unrelated to its active site, which can bind an effector molecule. This interaction is another mechanism of enzyme regulation. Allosteric modification usually happens in proteins with more than one subunit. Allosteric interactions are often present in metabolic pathways and are beneficial in that they allow one step of a reaction to regulate another step.[20] They allow an enzyme to have a range of molecular interactions, other than the highly specific active site.[20]
See also
References
- Stevenson, Robert Louis (1999). KIDNAPPED (1999 ed.). London: Fox Studio . p. 123.
- Bugg TD (2004). Introduction to Enzyme and Coenzyme Chemistry (PDF) (2nd ed.). Blackwell Publishing Limited. ISBN 9781405114523. Archived from the original (PDF) on 22 March 2018.
- Shanmugam S (2009). Enzyme Technology. I K International Publishing House. p. 48. ISBN 9789380026053.
- Pravda L, Berka K, Svobodová Vařeková R, et al. (2014). "Anatomy of Enzyme Channels". BMC Bioinformatics. 15 (1): 379. doi:10.1186/s12859-014-0379-x. PMC 4245731. PMID 25403510.
- Alberts B (2010). Essential Cell Biology. Garland Science. p. 91. ISBN 9780815341291.
- Dagmar R, Gregory A (2008). "How Enzymes Work". Science. 320 (5882): 1428–1429. doi:10.1126/science.1159747. PMID 18556536. S2CID 43617575.
- Robert A (2000). Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis (PDF) (2nd ed.). Wiley-Blackwell. ISBN 9780471359296.
- Kool ET (1984). "Active site tightness and substrate fit in DNA replication". Annual Review of Biochemistry. 71: 191–219. doi:10.1146/annurev.biochem.71.110601.135453. PMID 12045095.
- Csermely, Peter; Palotai, Robin; Nussinov, Ruth (2010). "Induced fit, conformational selection and independent dynamic segments: an extended view of binding events". Trends in Biochemical Sciences. 35 (10): 539–546. arXiv:1005.0348. doi:10.1016/j.tibs.2010.04.009. ISSN 0968-0004. PMC 3018770. PMID 20541943.
- Daniel E (1995). "The Key–Lock Theory and the Induced Fit Theory". Angewandte Chemie International Edition. 33 (2324): 2375–2378. doi:10.1002/anie.199423751.
- Sullivan SM (2008). "Enzymes with lid-gated active sites must operate by an induced fit mechanism instead of conformational selection". Proceedings of the National Academy of Sciences of the United States of America. 105 (37): 13829–13834. Bibcode:2008PNAS..10513829S. doi:10.1073/pnas.0805364105. PMC 2544539. PMID 18772387.
- Copeland, Robert A. (2013). "Drug–Target Residence Time". Evaluation of Enzyme Inhibitors in Drug Discovery. John Wiley & Sons, Ltd. pp. 287–344. ISBN 978-1-118-54039-8.
- Pan, Albert C.; Borhani, David W.; Dror, Ron O.; Shaw, David E. (2013). "Molecular determinants of drug–receptor binding kinetics". Drug Discovery Today. 18 (13–14): 667–673. doi:10.1016/j.drudis.2013.02.007. ISSN 1359-6446. PMID 23454741.
- Miller, Duncan C.; Lunn, Graham; Jones, Peter; Sabnis, Yogesh; Davies, Nichola L.; Driscoll, Paul (2012). "Investigation of the effect of molecular properties on the binding kinetics of a ligand to its biological target". MedChemComm. 3 (4): 449–452. doi:10.1039/c2md00270a. ISSN 2040-2503.
- Flexner C (1998). "HIV-protease inhibitors". The New England Journal of Medicine. 338 (18): 1281–1292. doi:10.1056/NEJM199804303381808. PMID 9562584.
- Ashraf B, Chi-Huey W (2003). "HIV-1 protease: mechanism and drug discovery". Organic & Biomolecular Chemistry. 1 (1): 5–14. doi:10.1039/B208248A. PMID 12929379.
- Gray W, Rick G (1993). "Cytoprotection by inhibition of chloride channels: The mechanism of action of glycine and strychnine". Life Sciences. 53 (15): 1211–1215. doi:10.1016/0024-3205(93)90539-F. PMID 8412478.
- Jansen EF, Nuttig F, Balls AK (1949). "Mode of inhibition of chymotrypsin by diisopropyl fluorophosphate; introduction of phosphorus". The Journal of Biological Chemistry. 179 (1): 201–204. doi:10.1016/S0021-9258(18)56828-9. PMID 18119235.
- Schechter I (2005). "Mapping of the active site of proteases in the 1960s and rational design of inhibitors/drugs in the 1990s". Current Protein & Peptide Science. 6 (6): 501–512. doi:10.2174/138920305774933286. PMID 16381600.
- DeDecker BS (2000). "Allosteric drugs: thinking outside the active-site box". Chemistry & Biology. 7 (5): 103–107. doi:10.1016/S1074-5521(00)00115-0. PMID 10801477.
- Zuercher M (2008). "Structure-Based Drug Design: Exploring the Proper Filling of Apolar Pockets at Enzyme Active Sites". Journal of Organic Chemistry. 73 (12): 4345–4361. doi:10.1021/jo800527n. PMID 18510366.
- Powers R (2006). "Comparison of protein active site structures for functional annotation of proteins and drug design". Proteins. 65 (1): 124–135. doi:10.1002/prot.21092. PMID 16862592. S2CID 2527166.
Further reading
- Alan Fersht, Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. W. H. Freeman, 1998. ISBN 0-7167-3268-8
- Bugg, T. Introduction to Enzyme and Coenzyme Chemistry. (2nd edition), Blackwell Publishing Limited, 2004. ISBN 1-4051-1452-5.