Galantamine total synthesis
The article concerns the total synthesis of galanthamine, a drug used for the treatment of mild to moderate Alzheimer's disease.[1]
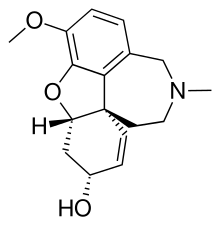
The natural source of galantamine are certain species of daffodil and because these species are scarce and because the isolation of galanthamine from daffodil is expensive (a 1996 figure specifies 50,000 US dollar per kilogram, the yield from daffodil is 0.1–0.2% dry weight) alternative synthetic sources are under development by means of total synthesis.
Outline
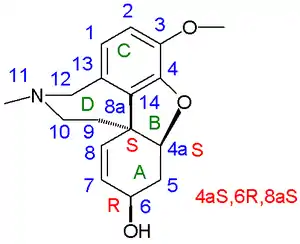
In 1962 racemic galanthamine and epi-galanthamine were prepared by organic reduction of racemic narwedine by D. H. R. Barton. Narwedine is the related enone (galanthamine the allyl alcohol) obtained in an oxidative coupling. Chemical yield: 1.4%. In addition they isolated (−)-narwardine by chiral resolution from a mixture of racemic narwedine and 0.5 equivalents of (+)-galanthamine. In this way they were able to obtain (−)galanthamine again by reduction In 1976 Kametani obtained both galanthamine enantiomers by using a derivative of tartaric acid as a chiral resolving agent. In 1977 Koga obtained both enantiomers via a chiral pool synthesis starting from L-tyrosine[2][3] and in 1988 Carrol optimized the oxidative coupling route to 11% yield based on isovanillin.
In 1989 Vlahov exploited asymmetric reduction by biocatalysis in the synthesis of several galanthamine precursors. and in 1994 Shieh & Carlson [4] obtained (−)-galanthamine by spontaneous resolution of its narwedine precursor. Racemic narwedine was treated with 0.01 equivalent of (+)-galanthamine resulting in a 76% yield. Narwedine is a racemic conglomerate allowing the isolation of the S,S enantiomer from the R,R enantiomer by simple crystallization. What made the process unique is that both enantiomers are in dynamic chemical equilibrium with each other through a common phenol in a Michael reaction-like reaction brought about by triethylamine.
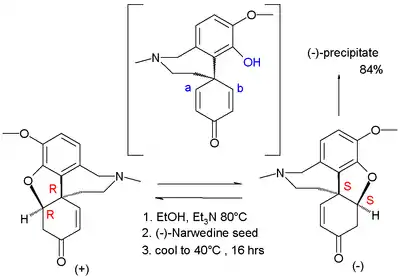 Resolution of Narwedine |
| Resolution of Narwedine |
|---|
In 1999 Jordis performed (−)-galanthamine synthesis on a multikilogram scale based on Carrol chemistry and Shieh/Carlson chiral resolution. This would become the basis for current industrial production by Sanochemia (AT). In 2000 Fels proposed an intramolecular Heck reaction for the construction of the galanthamine backbone and in the same year Trost & Toste obtained (−)-galanthamine in an asymmetric synthesis involving asymmetric allylic alkylation and an intramolecular Heck reaction. Improved methods were published in 2002 and 2005 (see below) In 2004 Node obtained (−)-galanthamine via a remote asymmetric induction method with starting chiral compound D-phenylalanine.[5] Brown prepared (−)-galanthamine in 2007 starting from isovanillin.[6] Isovanillin was also used by Magnus (2009) [7] D-glucose was used by Chida (2010).[8]
Syntheses of racemic galanthamine have been reported by Wang in 2006 [9] and by Saito in 2008.[10]
Sanochemia industrial production
The method outlined by Jordis in 1999 forms the basis for industrial galanthamine production.[11]
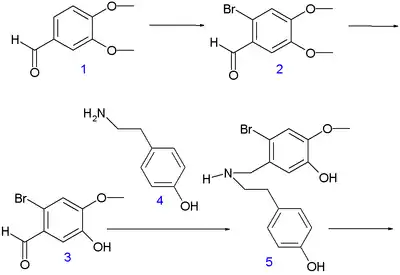 |
 | |
| Narwedine synthesis part A | Narwedine synthesis part B | |
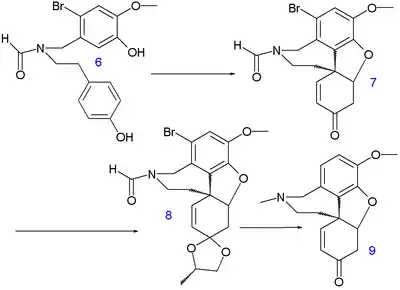
This method is based on electrophilic halogenation of 3,4-dimethoxybenzaldehyde 1 (accessible from isovanillin) with bromine / acetic acid to organobromide 2 followed by regioselective demethylation with sulfuric acid to phenol 3. This compound reacts in a reductive amination (sodium borohydride) with tyramine 4 to amine 5 which is formylated with ethyl formate and formic acid in dioxane in the next step to compound 6. An oxidative phenol coupling takes place next with Potassium ferricyanide and potassium carbonate in toluene to 7. The C8a-C14 bond is formed in the first step followed by a Michael addition of the other phenolic group to the newly formed enone group. The reaction step creates two stereocenters leading to two diastereomeric pairs of enantiomers. By the nature of the ABD skeleton the desired S,S/R,R pair is the major product formed and the other pair S,R/R,S is removed in workup. The ketone group is protected as the ketal 8 with 1,2-propylene glycol enabling the organic reduction by lithiumaluminiumhydride of both the bromine group and the formyl group. In the second phase the ketal group is removed (hydrochloric acid) forming racemic (S,S/R,R) narwedine 9.
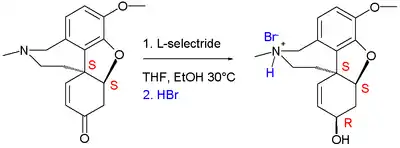
Enantiopure (−)-narwedine is obtained via the dynamic chiral resolution method pioneered by Shieh/Carlson and in the final step the ketone is reduced to the alcohol with L-selectride.
 reduction of (−)-narwedine to (−)-galanthamine as the bromide |
| reduction of (−)-narwedine to (−)-galanthamine as the bromide |
|---|
This final step is enantioselective producing the desired S,S,R compound because the approach of H− is restricted to the Si face as the Re face is shielded by the DB ring system. Formation of the S,S,S epimer is also avoided by keeping the reaction temperature below −15 °C.
Trost Galanthamine synthesis
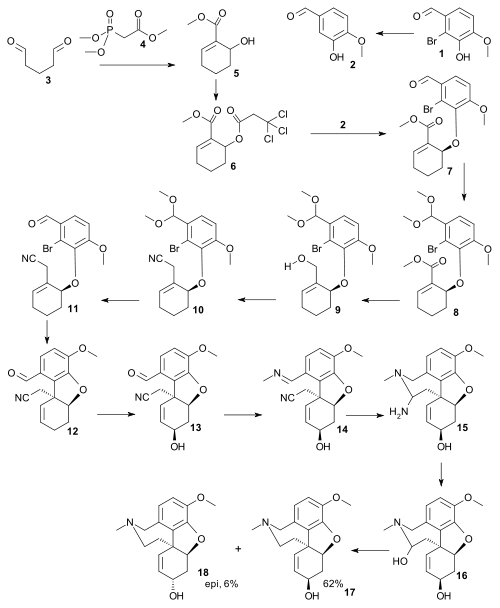
The total synthesis of galanthamine (Trost 2005) [12] is described as follows: the sequence starts by bromination by electrophilic aromatic substitution of isovanillin 1 to bromophenol 2, then by synthesis of the second intermediate 5 by reacting dialdehyde 3 in a coupled aldol reaction and Horner–Wadsworth–Emmons reaction with trimethyl phosphonoacetate 4. The hydroxyl group is activated as a trichloroethyl carbonate leaving group to 6. Next an enantioselective Trost AAA reaction takes place between bromophenol 2 and carbonate 6 to the allyl ether 7. Next the aldehyde group is protected as an acetal in 8 and this step enables the organic reduction of the ester group to the alcohol 9 with DIBAH and subsequent homologation of this alcohol to a nitrile by Mitsunobu-type reaction using acetone cyanohydrine as the source of cyanide, to yield 10 followed by aldehyde deprotection to 11. The intramolecular Heck reaction to 12 forms the dihydrofuran ring. Allylic oxidation by selenium dioxide provides allylic alcohol 13 with the correct stereochemistry. The aldehyde reacts with methylamine to the imine 14 and reduction of the imine and nitrile by DIBAL-H leading to ring-closure to the aminal 15 (not isolated) followed by acid quenching gives the hemi-aminal 16. In the final step the hemiaminal is reduced to give Galanthamine 17 together with 6% of the epi isomer 18.[13]
 Trost 2005 Galanthamine total synthesis |
| Trost 2005 Galanthamine total synthesis |
|---|
Eli Lilly / U. of Southampton Galanthamine synthesis
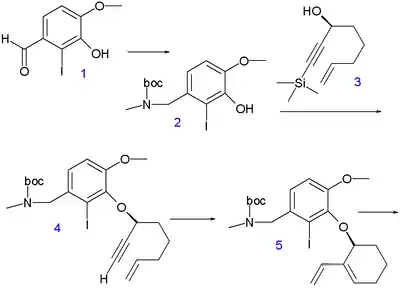
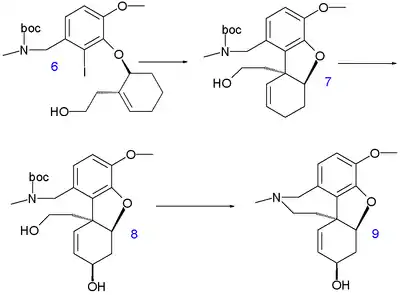
A total synthesis reported by Eli Lilly and the University of Southampton in 2007 also starts from isovanillin.[6] The aldehyde group in its derivative 1 is converted to its amine by reductive amination with methylamine which is then protected as a BOC group in 2. The remainder of the carbon framework is added with chiral propargyl alcohol 3 (introducing the 4a stereocenter and obtained by chiral synthesis of the ketone with R-Alpine borane) in a Mitsunobu reaction to aryl ether 4. The trimethylsilyl protective group is removed by potassium carbonate in methanol and the subsequent enyne metathesis reaction with Grubbs' catalyst gives diene 5. A hydroboration–oxidation reaction converts 5 to alcohol 6 and an intramolecular Heck reaction affords tricycle 7 with alkene isomerization and establishment of the 8a stereocenter with correct stereochemistry based on chiral induction. The allyl alcohol group in 8 is introduced by selenoxide oxidation with an excess of the desired diastereomer. In the final step to galanthamine 9 the hydroxyl group is activated as the triflate and the amine group as the mesylate for intramolecular azepine ring closure via nucleophilic substitution (with 6% epimer formation).
 |
 | |
| Galanthamine synthesis 2007 A | Galanthamine synthesis 2007 part B | |
References and notes
- Synthesis and Pharmacology of Galantamine José Marco-Contelles, Maria do Carmo Carreiras, Carolina Rodríguez, Mercedes Villarroya, and Antonio G. García Chem. Rev.; 2006; 106(1) pp 116–133; (Review) doi:10.1021/cr040415t
- Koga, Kenji; Tomioka, Kiyoshi; Shimizu, Kimihiro; Yamada, Shun-Ichi (1977). "Approaches to the Biogenetic-type Asymmetric Synthesis of Some Amaryllidaceae Alkaloids". Heterocycles. 6 (9): 1752. doi:10.3987/R-1977-09-1752.
- Koga, Kenji; Shimizu, Kimihiro; Tomioka, Kiyoshi; Yamada, Shun-Ichi (1977). "A Biogenetic-type Asymmetric Synthesis of Optically Active Amaryllidaceae Alkaloids: (+)- and (−)-Galanthamine from L-Tyrosine". Heterocycles. 8: 277. doi:10.3987/S(S)-1977-01-0277.
- Asymmetric Transformation of Either Enantiomer of Narwedine via Total Spontaneous Resolution Process, a Concise Solution to the Synthesis of (−)-Galanthamine Wen-Chung Shieh and John A. Carlson J. Org. Chem.; 1994; 59(18) pp 5463–5465; doi:10.1021/jo00097a060
- Kodama, Sumiaki; Hamashima, Yoshio; Nishide, Kiyoharu; Node, Manabu (2004). "Total Synthesis of (−)-Galanthamine by Remote Asymmetric Induction". Angewandte Chemie International Edition. 43 (20): 2659–2661. doi:10.1002/anie.200353636. PMID 18629982.
- Satcharoen, Vachiraporn; McLean, Neville J.; Kemp, Stephen C.; Camp, Nicholas P.; Brown, Richard C. D. (2007). "Stereocontrolled Synthesis of (−)-Galanthamine". Organic Letters. 9 (10): 1867–1869. doi:10.1021/ol070255i. PMID 17429978.
- Magnus, Philip; Sane, Neeraj; Fauber, Benjamin P.; Lynch, Vince (2009). "Concise Syntheses of (−)-Galanthamine and (±)-Codeine via Intramolecular Alkylation of a Phenol Derivative". Journal of the American Chemical Society. 131 (44): 16045–16047. doi:10.1021/ja9085534. PMID 19835379.
- Chida, Noritaka; Kato, Tomoaki; Yamada, Hisako (2010). "Total Synthesis of (+)- and (−)-Galanthamine". Heterocycles. 82: 563. doi:10.3987/COM-10-S(E)27. Archived from the original on 2011-07-22.
- Hu, Xiang-Dong; Tu, Yong Qiang; Zhang, En; Gao, Shuanhu; Wang, Shaohua; Wang, Aixia; Fan, Chun-An; Wang, Min (2006). "Total Synthesis of (±)-Galanthamine‡". Organic Letters. 8 (9): 1823–1825. doi:10.1021/ol060339b. PMID 16623560.
- Ishikawa, Teruhiko; Kudo, Kazuhiro; Kuroyabu, Ken; Uchida, Satoshi; Kudoh, Takayuki; Saito, Seiki (2008). "Domino Double Michael−Claisen Cyclizations: A Powerful General Tool for Introducing Quaternary Stereocenters at C(4) of Cyclohexane-1,3-diones and Total Synthesis of Diverse Families of Sterically Congested Alkaloids". The Journal of Organic Chemistry. 73 (19): 7498–7508. doi:10.1021/jo801316s. PMID 18781800.
- Development of a Pilot Scale Process for the Anti-Alzheimer Drug (−)-Galanthamine Using Large-Scale Phenolic Oxidative Coupling and Crystallisation-Induced Chiral Conversion Bernhard Küenburg, Laszlo Czollner, Johannes Fröhlich, and Ulrich Jordis Org. Process Res. Dev.; 1999; 3(6) pp 425–431; (Article) doi:10.1021/op990019q
- Divergent Enantioselective Synthesis of (−)-Galanthamine and (−)-Morphine Barry M. Trost, Weiping Tang, and F. Dean Toste J. Am. Chem. Soc.; 2005; 127(42) pp 14785–14803; (Article) doi:10.1021/ja054449+
- a bromine, sodium acetate, acetic acid, iron, rt b potassium carbonate, 2days c Troc-Cl, DMAP, Pyridine, dichloromethane d palladium, Trost ligand, triethylamine, dichloromethane e 1.5 mol % TsOH, CH(OMe)3, methanol f DIBAL-H, toluene, −78 °C, 1 hr g triphenylphosphine, acetonecyanohydrin, DIAD, diethyl ether h 2.20 mol % TsOH, THF, water i 15 mol % Palladium(II) acetate, 15 mol% dppp, 3 eq. Ag2CO3, toluene, 107 °C j selenium dioxide disodium phosphate dioxane, 150 °C 3 hrs k methylamine, methanol l 4 eq. DIBAL-H, m aqueous NaH2PO4 n NaCNBH3