Carcinoid
A carcinoid (also carcinoid tumor) is a slow-growing[1] type of neuroendocrine tumor originating in the cells of the neuroendocrine system. In some cases, metastasis may occur. Carcinoid tumors of the midgut (jejunum, ileum, appendix, and cecum) are associated with carcinoid syndrome.
| Carcinoid | |
|---|---|
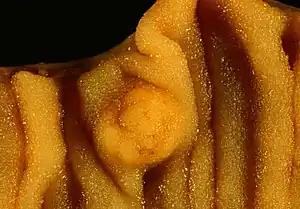 | |
| Picture of a carcinoid tumor (center of image) that encroaches into the lumen of the small bowel (pathology specimen). The prominent folds are plicae circulares, a characteristic of the small bowel. | |
| Specialty | Oncology |
Sometimes, carcinoids cause paraneoplastic syndromes, which involve discharge of serotonin and other vasoactive substances from well-differentiated carcinoids.[2][3] A neuroendocrine paraneoplastic syndrome involves neoplastic secretion of functional peptides, hormones, cytokines, growth factors, and/or immune cross-reactivity between tumor tissues and normal host tissues, resulting in a syndrome of clinical signs and symptoms.[4]
Carcinoid tumors are the most common malignant tumor of the appendix, but they are most commonly associated with the small intestine, and they can also be found in the rectum and stomach. They are known to grow in the liver, but this finding is usually a manifestation of metastatic disease from a primary carcinoid occurring elsewhere in the body. They have a very slow growth rate compared to most malignant tumors. The median age at diagnosis for all patients with neuroendocrine tumors is 63 years.
Signs and symptoms
While most carcinoids are asymptomatic through the natural life and are discovered only upon surgery for unrelated reasons (so-called coincidental carcinoids), all carcinoids are considered to have malignant potential.
About 10% of carcinoids secrete excessive levels of a range of hormones, most notably serotonin (5-hydroxytryptamine), causing:
- Flushing (serotonin itself does not cause flushing). Potential causes of flushing in carcinoid syndrome include bradykinins, prostaglandins, tachykinins, substance P, and/or histamine, diarrhea, and heart problems. Because of serotonin's growth-promoting effect on cardiac myocytes, a serotonin-secreting carcinoid tumour may cause a tricuspid valve disease syndrome, due to the proliferation of myocytes onto the valve.[5]
- Diarrhea
- Wheezing
- Abdominal cramping
- Peripheral edema
The outflow of serotonin can cause a depletion of tryptophan leading to niacin deficiency. Niacin deficiency, also known as pellagra, is associated with dermatitis, dementia, and diarrhea.
This constellation of symptoms is called carcinoid syndrome or (if acute) carcinoid crisis. Occasionally, haemorrhage or the effects of tumor bulk are the presenting symptoms. The most common originating site of carcinoid is the small bowel, particularly the ileum; carcinoid tumors are the most common malignancy of the appendix. Carcinoid tumors may rarely arise from the ovary or thymus.[6]
They are most commonly found in the midgut at the level of the ileum or in the appendix. The next most commonly affected area is the respiratory tract, with 28% of all cases—per PAN-SEER data (1973–1999). The rectum is also a common site.
Gastrointestinal
Carcinoid tumors are apudomas that arise from the enterochromaffin cells throughout the gut. Over two-thirds of carcinoid tumors are found in the gastrointestinal tract.[7]
Lung
Carcinoid tumors are also found in the lungs.
Other sites and metastases
Metastasis of carcinoid can lead to carcinoid syndrome. This is due to the over-production of many substances, including serotonin, which are released into the systemic circulation, and which can lead to symptoms of cutaneous flushing, diarrhea, bronchoconstriction, and right-sided cardiac valve disease. It is estimated that less than 6% of carcinoid patients will develop carcinoid syndrome, and of these, 50% will have cardiac involvement.[8]
Goblet cell carcinoid
This is considered to be a hybrid between an exocrine and endocrine tumor derived from crypt cells of the appendix. Histologically, it forms clusters of goblet cells containing mucin with a minor admixture of Paneth cells and endocrine cells. The growth pattern is distinctive: typically producing a concentric band of tumor nests interspersed among the muscle and stroma of the appendiceal wall extending up the shaft of the appendix. This makes the lesion difficult to suspect grossly and difficult to measure. Small tumor nests may be camouflaged amongst the muscle or in periappendiceal fat; cytokeratin preparations best demonstrate the tumor cells; mucin stains are also helpful in identifying them. They behave in a more aggressive manner than do classical appendiceal carcinoids. Spread is usually to regional lymph nodes, peritoneum, and particularly the ovary. They do not produce sufficient hormonal substances to cause carcinoid or other endocrine syndromes. In fact, they more closely resemble exocrine than endocrine tumors. The term 'crypt cell carcinoma' has been used for them, and though perhaps more accurate than considering them carcinoids, has not been a successful competitor.
Cause
Carcinoid syndrome involves multiple tumors in one out of five males. The incidence of gastric carcinoids is increased in achlorhydria, Hashimoto's thyroiditis, and pernicious anemia.
Treatment
Surgery, if feasible, is the only curative therapy. If the tumor has metastasized (most commonly, to the liver) and is considered incurable, there are some promising treatment modalities, such as the radiopharmaceuticals Lutetium (177Lu) DOTA-octreotate) and 131I-mIBG (meta iodo benzyl guanidine[9]) for arresting the growth of the tumors and prolonging survival in patients with liver metastases, though these are currently experimental.
Chemotherapy is of little benefit and is generally not indicated. Octreotide or lanreotide (somatostatin analogues) may decrease the secretory activity of the carcinoid, and may also have an anti-proliferative effect. Interferon treatment is also effective, and usually combined with somatostatin analogues.
As the metastatic potential of a coincidental carcinoid is probably low, the current recommendation is for follow up in 3 months with CT or MRI, labs for tumor markers such as serotonin, and a history and physical, with annual physicals thereafter.
History
They were first characterized in 1907 by Siegfried Oberndorfer, a German pathologist at the University of Munich, who coined the term karzinoide, or "carcinoma-like", to describe the unique feature of behaving like a benign tumor despite having a malignant appearance microscopically. The recognition of their endocrine-related properties was later described by Gosset and Masson in 1914, and these tumors are now known to arise from the enterochromaffin (EC) and enterochromaffin-like (ECL) cells of the gut. Some sources credit Otto Lubarsch with the discovery.[10]
In 2000, the World Health Organization redefined "carcinoid", but this new definition has not been accepted by all practitioners.[11] This has led to some complexity in distinguishing between carcinoid and other neuroendocrine tumors in the literature. According to the American Cancer Society, the 2000 WHO definition states:[11]
The WHO now divides these growths into neuroendocrine tumors and neuroendocrine cancers. Neuroendocrine tumors are growths that look benign but that might possibly be able to spread to other parts of the body. Neuroendocrine cancers are abnormal growths of neuroendocrine cells which can spread to other parts of the body.
Other organisms
Specific to rodents, chronically reduced gastric acid secretion encourages carcinoid development.[12]: 812
See also
- Carcinoid syndrome
- Don Meyer, head coach emeritus of the Northern State University men's basketball team. Meyer was found to have carcinoid cancer following an automobile accident in September 2009.
- Derrick Bell, professor and legal scholar, died of carcinoid cancer on October 5, 2011.
References
- Maroun J, Kocha W, Kvols L, et al. (April 2006). "Guidelines for the diagnosis and management of carcinoid tumors. Part 1: The gastrointestinal tract. A statement from a Canadian National Carcinoid Expert Group". Curr Oncol. 13 (2): 67–76. doi:10.3390/curroncol13020006. PMC 1891174. PMID 17576444.
- Gade, Ajay K.; Olariu, Eva; Douthit, Nathan T.; Gade, Ajay K.; Olariu, Eva; Douthit, Nathan T. (2020-03-05). "Carcinoid Syndrome: A Review". Cureus. 12 (3): e7186. doi:10.7759/cureus.7186. ISSN 2168-8184. PMC 7124884. PMID 32257725.
- Tsoli, Marina; Dimitriadis, Georgios K.; Androulakis, Ioannis I.; Kaltsas, Gregory; Grossman, Ashley (2000). Feingold, Kenneth R.; Anawalt, Bradley; Blackman, Marc R.; Boyce, Alison (eds.). Paraneoplastic Syndromes Related to Neuroendocrine Tumors. PMID 25905358. Retrieved 2023-02-19.
{{cite book}}:|work=ignored (help) - Guilmette, Julie; Nosé, Vânia (2019-07-01). "Paraneoplastic syndromes and other systemic disorders associated with neuroendocrine neoplasms". Seminars in Diagnostic Pathology. 36 (4): 229–239. doi:10.1053/j.semdp.2019.03.002. ISSN 0740-2570. PMID 30910348. S2CID 85514650.
- "Carcinoid Tumors and Syndrome". The Lecturio Medical Concept Library. Retrieved 5 July 2021.
- Daffner KR, Sherman JC, Gilberto Gonzalez R, Hasserjian RP (2008). "Case 35-2008 — A 65-Year-Old Man with Confusion and Memory Loss". N Engl J Med. 359 (20): 2155–2164. doi:10.1056/NEJMcpc0804643. PMID 19005200.
- Modlin IM, Lye KD, Kidd M (February 2003). "A 5-decade analysis of 13,715 carcinoid tumors". Cancer. 97 (4): 934–59. doi:10.1002/cncr.11105. PMID 12569593.
- Fox DJ, Khattar RS (2004). "Carcinoid heart disease: presentation, diagnosis, and management". Heart. 90 (10): 1224–8. doi:10.1136/hrt.2004.040329. PMC 1768473. PMID 15367531.
- "Medical Reviews | The Carcinoid Cancer Foundation". 2015-11-15. Archived from the original on 2015-11-15. Retrieved 2023-09-11.
- Kulke MH, Mayer RJ (March 1999). "Carcinoid tumors". N. Engl. J. Med. 340 (11): 858–68. doi:10.1056/NEJM199903183401107. PMID 10080850.
- "ACS :: What Is a Gastrointestinal Carcinoid Tumor?". Archived from the original on 2016-11-05. Retrieved 2011-06-02.
- Haschek, Wanda; Rousseaux, Colin; Wallig, Matthew (2013). Haschek and Rousseaux's Handbook of Toxicologic Pathology. Academic Press. doi:10.1016/c2010-1-67850-9. ISBN 978-0-12-415759-0.