Gastrointestinal stromal tumor
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of the gastrointestinal tract. GISTs arise in the smooth muscle pacemaker interstitial cell of Cajal, or similar cells.[2] They are defined as tumors whose behavior is driven by mutations in the KIT gene (85%),[2] PDGFRA gene (10%),[2] or BRAF kinase (rare).[2] 95% of GISTs stain positively for KIT (CD117).[2][3] Most (66%) occur in the stomach and gastric GISTs have a lower malignant potential than tumors found elsewhere in the GI tract.[3]
| Gastrointestinal stromal tumor | |
|---|---|
.jpg.webp) | |
| Histopathologic image of gastrointestinal stromal tumor of the stomach. Hematoxylin-eosin stain. | |
| Specialty | Oncology |
| Differential diagnosis | Ectopic pancreas[1] |
Classification
GIST was introduced as a diagnostic term in 1983.[2]: 1060 Until the late 1990s, many non-epithelial tumors of the gastrointestinal tract were called "gastrointestinal stromal tumors". Histopathologists were unable to specifically distinguish among types now known to be dissimilar molecularly. Subsequently, CD34, and later CD117, were identified as markers that could distinguish the various types. Additionally, in the absence of specific therapy, the diagnostic categorization had only a limited influence on prognosis and therapy.
The understanding of GIST biology changed significantly with identification of the molecular basis of GIST,[2]: 1065 particularly c-KIT. Historically, literature reviews prior to the molecular definition of GIST, and for a short time thereafter, asserted that 70-80% of GISTs were benign.[4][5][6] The identification of a molecular basis for GIST led to the exclusion of many tumors that had been considered as GIST previously, and also the incorporation of a much larger number of tumors that had been labeled as other types of sarcomas and undifferentiated carcinomas.[2]: 1065 For example, some previous diagnoses of stomach and small bowel leiomyosarcomas (malignant tumor of smooth muscle) would be reclassified as GISTs on the basis of immunohistochemical staining. All GIST tumors are now considered to have malignant potential, and no GIST tumor can be definitively classified as "benign".[7] Hence, all GISTs are eligible for cancer staging in the AJCC (7th edition) / UICC.[8] Nonetheless, different GISTs have different risk assessments of their tendency to recur or to metastasize, dependent on their site of origin, size, and number of mitotic figures.
Due to the change in definition, clinical pathways of care before the year 2000 are largely uninformative in the current era.[2]
Signs and symptoms
GISTs may present with trouble swallowing, gastrointestinal bleeding, or metastases (mainly in the liver). Intestinal obstruction is rare, due to the tumor's outward pattern of growth. Often, there is a history of vague abdominal pain or discomfort, and the tumor has become rather large by time the diagnosis is made.
Pathophysiology
GISTs are tumors of connective tissue, i.e. sarcomas; unlike most gastrointestinal tumors, they are nonepithelial. About 70% occur in the stomach, 20% in the small intestine and less than 10% in the esophagus. Small tumors are generally not aggressive, especially when cell division rate is slow. GIST tumors commonly metastasize to the liver (in 28% of cases) and/or to the greater omentum, lesser omentum, or mesentery (in 30% of cases). Less common areas of metastasis include the lungs, subcutaneous tissue, lymph nodes or bones.[9]
GISTs are thought to arise from interstitial cells of Cajal (ICC), that are normally part of the autonomic nervous system of the intestine.[3] They serve a pacemaker function in controlling motility.
Genetics
Most GISTs are sporadic. Less than 5% occur as part of hereditary familial or idiopathic multitumor syndromes. These include, in descending order of frequency, neurofibromatosis Recklinghausen (NF-1), Carney's triad (gastric GIST, pulmonary chondroma and extra-adrenal paraganglioma), germline gain-of-function mutations in c-KIT/PDGFRA, and the Carney-Stratakis syndrome.[10] The Carney-Stratakis syndrome is a dyad of hereditary GIST and paraganglioma, that is caused by germline mutations in the mitochondrial tumor suppressor gene pathway involving the succinate dehydrogenase (SDH) subunits SDHD, SDHC and SDHB.[11]
c-KIT mutations
Approximately 85% GISTs are associated with an abnormal c-KIT pathway. c-KIT is a gene that encodes for a transmembrane receptor for a growth factor termed stem cell factor (scf). The abnormal c-KIT pathway most commonly (85%) arises from mutation of the gene itself; a smaller subset of c-KIT-associated GISTs are associated with constitutive activity of the KIT enzymatic pathway, found by immunoblotting.[2]: 1062 The c-KIT product/CD117 is expressed on ICCs and a large number of other cells, mainly bone marrow cells, mast cells, melanocytes and several others. In the gut, however, a mass staining positive for CD117 is likely to be a GIST, arising from ICC cells.
The c-KIT molecule comprises a long extracellular domain, a transmembrane segment, and an intracellular part. Mutations generally occur in the DNA encoding the intracellular part (exon 11), which acts as a tyrosine kinase to activate other enzymes. Mutations make c-KITfunction independent of activation by scf, leading to a high cell division rate and possibly genomic instability. Additional mutations are likely "required" for a cell with a c-KIT mutation to develop into a GIST, but the c-KIT mutation is probably the first step of this process.
Mutations in the exons 11, 9 and rarely 13 and 17 of the c-KIT gene are known to occur in GIST. The tyrosine kinase function of c-KIT is important in the medical therapy for GISTs, as described below.
- KIT-D816V point mutations in c-KIT exon 17 are responsible for resistance to targeted therapy drugs like imatinib mesylate, a tyrosine kinase inhibitor.
- KIT-p.D419del (exon 8) — A subset of gastrointestinal stromal tumors previously regarded as wild-type tumors carries somatic activating mutations in KIT exon 8 (p.D419del).[12]
PDGFRA mutations
Most GIST cells with wildtype (i.e. not mutated) c-KIT instead have a mutation in another gene, PDGFR-α (platelet-derived growth factor receptor alpha), which is a related tyrosine kinase. Mutations in c-KIT and PDGFrA are mutually exclusive .
Wild-type tumors
Lesser numbers of GISTs appear to be associated with neither c-KIT nor PDGFR-α abnormalities.[2]: 1062 About 10-15% of gastrointestinal stromal tumors (GISTs) carry wild-type sequences in all hot spots of KIT and platelet-derived growth factor receptor alpha (PDGFRA) (wt-GISTs). These tumors are currently defined by having no mutations in exons 9, 11, 13, and 17 of the KIT gene and exons 12, 14, and 18 of the PDGFRA gene.[12]
Diagnosis

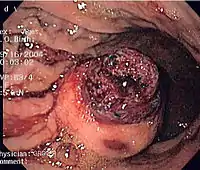
CT scanning is often undertaken (see the radiology section).
The definitive diagnosis is made with a biopsy, which can be obtained endoscopically, percutaneously with CT or ultrasound guidance or at the time of surgery. A biopsy sample will be investigated under the microscope by a pathologist physician. The pathologist examines the histopathology to identify the characteristics of GISTs (spindle cells in 70-80%, epitheloid aspect in 20-30%). Smaller tumors can usually be confined to the muscularis propria layer of the intestinal wall. Large ones grow, mainly outward, from the bowel wall until the point where they outstrip their blood supply and necrose (die) on the inside, forming a cavity that may eventually come to communicate with the bowel lumen.
When GIST is suspected—as opposed to other causes for similar tumors—the pathologist can use immunohistochemistry (specific antibodies that stain the molecule CD117 [also known as c-KIT] —see below). 95% of all GISTs are CD117-positive (other possible markers include CD34, DOG-1, desmin, and vimentin). Other cells that show CD117 positivity are mast cells.
If the CD117 stain is negative and suspicion remains that the tumor is a GIST, the newer antibody DOG-1 (Discovered On GIST-1) can be used. Also, sequencing of KIT and PDGFRA can be used to prove the diagnosis.
Imaging
The purpose of radiologic imaging is to locate the lesion, evaluate for signs of invasion and detect metastasis. Features of GIST vary depending on tumor size and organ of origin. The diameter can range from a few millimeters to more than 30 cm. Larger tumors usually cause symptoms in contrast to those found incidentally which tend to be smaller and have better prognosis.[4][13] Large tumors tend to exhibit malignant behavior but small GISTs may also demonstrate clinically aggressive behavior.[14]

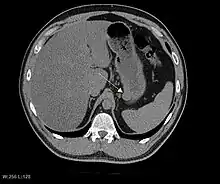
Plain radiographs are not very helpful in the evaluation of GISTs. If an abnormality is seen, it will be an indirect sign due to the tumor mass effect on adjacent organs. On abdominal x-ray, stomach GISTs may appear as a radiopaque mass altering the shape of the gastric air shadow. Intestinal GISTs may displace loops of bowel and larger tumors may obstruct the bowel and films will show an obstructive pattern. If cavitations are present, plain radiographs will show collections of air within the tumor.[15] Calcification is an unusual feature of GIST but if present can be visible on plain films.
Barium fluoroscopic examinations and CT are commonly used to evaluate the patient with abdominal complaints. Barium swallow images show abnormalities in 80% of GIST cases.[14] However, some GISTs may be located entirely outside the lumen of the bowel and will not be appreciated with a barium swallow. Even in cases when the barium swallow is abnormal, an MRI or CT scan must follow since it is impossible to evaluate abdominal cavities and other abdominal organs with a barium swallow alone. In a CT scan, abnormalities may be seen in 87% of patients and it should be made with both oral and intravenous contrast.[14] Among imaging studies, MRI has the best tissue contrast, which aids in the identification of masses within the GI tract (intramural masses). Intravenous contrast material is needed to evaluate lesion vascularity.
Preferred imaging modalities in the evaluation of GISTs are CT and MRI,[16]: 20–21 and, in selected situations, endoscopic ultrasound. CT advantages include its ability to demonstrate evidence of nearby organ invasion, ascites, and metastases. The ability of an MRI to produce images in multiple planes is helpful in determining the bowel as the organ of origin (which is difficult when the tumor is very large), facilitating diagnosis.
Small GISTs
Since GISTs arise from the bowel layer called muscularis propria (which is deeper to the mucosa and submucosa from a luminal perspective), small GIST imaging usually suggest a submucosal process or a mass within the bowel wall. In barium swallow studies, these GISTs most commonly present with smooth borders forming right or obtuse angles with the nearby bowel wall, as seen with any other intramural mass. The mucosal surface is usually intact except for areas of ulceration, which are generally present in 50% of GISTs. Ulcerations fill with barium causing a bull's eye or target lesion appearance. In contrast-enhanced CT, small GISTs are seen as smooth, sharply defined intramural masses with homogeneous attenuation.
Large GISTs
As the tumor grows it may project outside the bowel (exophytic growth) and/or inside the bowel (intraluminal growth), but they most commonly grow exophytically such that the bulk of the tumor projects into the abdominal cavity. If the tumor outstrips its blood supply, it can necrose internally, creating a central fluid-filled cavity with bleeding and cavitations that can eventually ulcerate and communicate into the lumen of the bowel. In that case, barium swallow may show an air, air-fluid levels or oral contrast media accumulation within these areas.[14][17] Mucosal ulcerations may also be present. In contrast-enhanced CT images, large GISTs appear as heterogeneous masses due to areas of living tumor cells surrounding bleeding, necrosis or cysts, which is radiographically seen as a peripheral enhancement pattern with a low attenuation center.[13] In MRI studies, the degree of necrosis and bleeding affects the signal intensity pattern. Areas of bleeding within the tumor will vary its signal intensity depending on how long ago the bleeding occurred. The solid portions of the tumor are typically low signal intensity on T1-weighted images, are high signal intensity on T2-weighted images and enhanced after administration of gadolinium. Signal-intensity voids are present if there is gas within areas of necrotic tumor.[15][18][19]
Features of malignancy
Malignancy is characterized by local invasion and metastases, usually to the liver, omentum and peritoneum. However, cases of metastases to bone, pleura, lungs and retroperitoneum have been seen. In distinction to gastric adenocarcinoma or gastric/small bowel lymphoma, malignant lymphadenopathy (swollen lymph nodes) is uncommon (<10%) and thus imaging usually shows absence of lymph node enlargement.[13] If metastases are not present, other radiologic features suggesting malignancy include: size (>5 cm), heterogeneous enhancement after contrast administration, and ulcerations.[4][13][20] Also, overtly malignant behavior (in distinction to malignant potential of lesser degree) is less commonly seen in gastric tumors, with a ratio of behaviorally benign to overtly malignant of 3-5:1.[4] Even if radiographic malignant features are present, these findings may also represent other tumors and definitive diagnosis must be made immunochemically.
Management

For localized, resectable adult GISTs, if anatomically and physiologically feasible, surgery is the primary treatment of choice.[16]: 69 Surgery can be potentially curative, but watchful waiting may be considered in small tumors in carefully selected situations.[21] Post-surgical adjuvant treatment may be recommended.[22] Lymph node metastases are rare, and routine removal of lymph nodes is typically not necessary. Laparoscopic surgery, a minimally invasive abdominal surgery using telescopes and specialized instruments, has been shown to be effective for removal of these tumors without needing large incisions.[23] The clinical issues of exact surgical indications for tumor size are controversial. The decision of appropriate laparoscopic surgery is affected by tumor size, location, and growth pattern.[24]
Radiotherapy has not historically been effective for GISTs[25]: 1122 and GISTs do not respond to most chemotherapy medications,[25]: 1122 with responses in less than 5%.[2]: 1065 However, several medications have been identified for clinical benefit in GIST: imatinib, sunitinib, regorafenib, ripretinib and avapritinib.
Imatinib (Glivec/Gleevec), an orally administered drug initially marketed for chronic myelogenous leukemia based on bcr-abl inhibition, also inhibits both c-KIT tyrosine kinase mutations and PDGFRA mutations other than D842V, and is useful in treating GISTs in several situations. Imatinib has been used in selected neoadjuvant settings.[26][16]: 23 In the adjuvant treatment setting, the majority of GIST tumors are cured by surgery, and do not need adjuvant therapy.[27] [28] An exception to this is where the anatomical position of the tumour means that surgery is technically difficult or complex. For example, rectal GIST often requires radical surgery to achieve complete resection, involving abdominoperineal resection and permanent stoma. In these situations, the use of neoadjuvant imatinib can significantly decrease both tumour size and mitotic activity, and permit less radical sphincter-preserving surgery.[26]
A substantial proportion of GIST tumors have a high risk of recurrence as estimated by a number of validated risk stratification schemes, and can be considered for adjuvant therapy.[28][29] The selection criteria underpinning the decision for possible use of imatinib in these settings, including a risk assessment based on pathological factors such as tumor size, mitotic rate and location, can be used to predict the risk of recurrence in GIST patients. Tumors <2 cm with a mitotic rate of <5/50 HPF have been shown to have lower risk of recurrence than larger or more aggressive tumors. Following surgical resection of GISTs, adjuvant treatment with imatinib reduces the risk of disease recurrence in higher risk groups. In selected higher risk adjuvant situations, imatinib is recommended for 3 years.[30]
Imatinib was approved for metastatic and unresectable GIST by the US FDA, February 1, 2002. The two-year survival of patients with advanced disease has risen to 75–80% following imatinib treatment.[31]
If resistance to imatinib is encountered, the multiple tyrosine kinase inhibitor sunitinib (marketed as Sutent) can be considered.[16]: 26 and 31 [32]
The effectiveness of imatinib and sunitinib depend on the genotype.[33] c-KIT- and PDGFRA-mutation negative GIST tumors are usually resistant to treatment with imatinib,[11] as is neurofibromatosis-1-associated wild-type GIST.[28] A specific subtype of PDGFRA mutation, D842V, is also insensitive to imatinib.[28][34] Recently, in PDGFRA-mutated GIST, avapritinib has been approved by FDA.[35] Now there are real-world data coming for avapritinib as well (Dr Sameer Rastogi, et al.)[36]
Regorafenib (Stivarga) was FDA-approved in 2013 for advanced GISTs that cannot be surgically removed and that no longer respond to imatinib (Gleevec) and sunitinib (Sutent).[37]
Epidemiology
GISTs occur in 10-20 per one million people. The true incidence might be higher, as novel laboratory methods are much more sensitive in diagnosing GISTs. The estimated incidence of GIST in the United States is approximately 5000 cases annually.[2]: 1063 This makes GIST the most common form of sarcoma, which constitutes more than 70 types of cancer.
The majority of GISTs present at ages 50–70 years. Across most of the age spectrum, the incidence of GIST is similar in men and women.[25]: 1122
Adult GISTs are rare before age 40. Pediatric GISTs are considered to be biologically distinct.[38] Unlike GISTs at other ages, pediatric GISTs are more common in girls and young women. They appear to lack oncogenic activating tyrosine kinase mutations in both KIT and PDGFRA.[39] Pediatric GISTs are treated differently from adult GISTs. Although the generally accepted definition of pediatric GIST is a tumor that is diagnosed at the age of 18 years or younger,[38] "pediatric-type" GISTs can be seen in adults, which affects risk assessment, the role of lymph node resection, and choice of therapy.[40]
Citations
- Yuan, Z; Chen, J; Zheng, Q; Huang, XY; Yang, Z; Tang, J (7 August 2009). "Heterotopic pancreas in the gastrointestinal tract". World Journal of Gastroenterology. 15 (29): 3701–3. doi:10.3748/wjg.15.3701. PMC 2721251. PMID 19653355.
- Demetri, G. (2011). "Gastrointestinal Stromal Tumor". In DeVita, L; Lawrence, TS; Rosenberg, SA (eds.). DeVita, Hellman, and Rosenberg's Cancer: Principles and Practice of Oncology (9th ed.). Wolters Kluwer Health/Lippincott Williams & Wilkins. ISBN 978-1-4511-0545-2.
- Miettinen M, Lasota J (2006). "Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis". Arch Pathol Lab Med. 130 (10): 1466–78. doi:10.5858/2006-130-1466-GSTROM. PMID 17090188.
- Burkill GJ, Badran M, Al-Muderis O, Meirion Thomas J, Judson IR, Fisher C, Moskovic EC (2003). "Malignant gastrointestinal stromal tumor: distribution, imaging features, and pattern of metastatic spread". Radiology. 226 (2): 527–32. doi:10.1148/radiol.2262011880. PMID 12563150.
- Nishida T, Hirota S (2000). "Biological and clinical review of stromal tumors in the gastrointestinal tract". Histol Histopathol. 15 (4): 1293–301. PMID 11005253.
- Miettinen M, Lasota J (2001). "Gastrointestinal stromal tumors--definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis". Virchows Arch. 438 (1): 1–12. doi:10.1007/s004280000338. PMID 11213830. S2CID 7598241.
- Raut, Chandrajit; Dematteo, Ronald (March 2008). "Evidence-Guided Surgical Management of GIST: Beyond a Simple Case of Benign and Malignant". Ann. Surg. Oncol. 15 (5): 1542–1543. doi:10.1245/s10434-008-9817-1. S2CID 12586147.
- AJCC manual
- Parab, Trisha M.; DeRogatis, Michael J.; Boaz, Alexander M.; Grasso, Salvatore A.; Issack, Paul S.; Duarte, David A.; Urayeneza, Olivier; Vahdat, Saloomeh; Qiao, Jian-Hua; Hinika, Gudata S. (February 2019). "Gastrointestinal stromal tumors: a comprehensive review". Journal of Gastrointestinal Oncology. 10 (1): 144–154. doi:10.21037/jgo.2018.08.20. PMC 6351301. PMID 30788170.
- Agaimy A, Hartmann A (2010). "Hereditary and non-hereditary syndromic gastointestinal stromal tumours". Pathologe (in German). 31 (6): 430–7. doi:10.1007/s00292-010-1354-6. PMID 20848108. S2CID 9295361.
- Stratakis CA, Carney JA (Jul 2009). "The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): molecular genetics and clinical implications". J Intern Med. 266 (1): 43–52. doi:10.1111/j.1365-2796.2009.02110.x. PMC 3129547. PMID 19522824.
- Huss, S; Künstlinger, H; Wardelmann, E; Kleine, M. A.; Binot, E; Merkelbach-Bruse, S; Rüdiger, T; Mittler, J; Hartmann, W; Büttner, R; Schildhaus, H. U. (2013). "A subset of gastrointestinal stromal tumors previously regarded as wild-type tumors carries somatic activating mutations in KIT exon 8 (p.D419del)". Modern Pathology. 26 (7): 1004–12. doi:10.1038/modpathol.2013.47. PMC 3701292. PMID 23599150.
- Hersh MR, Choi J, Garrett C, Clark R (2005). "Imaging Gastrointestinal Stromal Tumors". Cancer Control. 12 (2): 111–115. doi:10.1177/107327480501200206. PMID 15855894. S2CID 26071847.
- Pidhorecky I, Cheney RT, Kraybill WG, Gibbs JF (2000). "Gastrointestinal stromal tumors: current diagnosis, biologic behavior, and management". Ann Surg Oncol. 7 (9): 705–12. doi:10.1007/s10434-000-0705-6. PMID 11034250. S2CID 663887.
- Shojaku H, Futatsuya R, Seto H, et al. (1997). "Malignant gastrointestinal stromal tumor of the small intestine: radiologic-pathologic correlation". Radiat Med. 15 (3): 189–92. PMID 9278378.
- NCCN Clinical Practice Guidelines in Oncology Soft Tissue Sarcomas, version 3.2012. National Comprehensive Cancer Network.
- Lehnert T (1998). "Gastrointestinal sarcoma (GIST)--a review of surgical management". Ann Chir Gynaecol. 87 (4): 297–305. PMID 9891770.
- Levine MS, Buck JL, Pantongrag-Brown L, et al. (1996). "Leiomyosarcoma of the esophagus: radiographic findings in 10 patients". AJR Am J Roentgenol. 167 (1): 27–32. doi:10.2214/ajr.167.1.8659399. PMID 8659399.
- Tervahartiala P, Halavaara J (1998). "Radiology of GIST. Gastrointestinal stromal tumours". Ann Chir Gynaecol. 87 (4): 291–2. PMID 9891768.
- Ulusan S, Koc Z, Kayaselcuk F (2008). "Gastrointestinal stromal tumours: CT findings". Br J Radiol. 81 (968): 618–623. doi:10.1259/bjr/90134736. PMID 18628330.
- Casali PG, Blay JY (2010). "Gastrointestinal stromal tumours: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up". Annals of Oncology. 21 (suppl 5): v98–v102. doi:10.1093/annonc/mdq208. PMID 20555113.
- Bamboat ZM (2012). "Updates on the management of gastrointestinal stromal tumors". Surg Oncol Clin N Am. 21 (2): 301–16. doi:10.1016/j.soc.2011.12.004. PMC 3386646. PMID 22365521.
- Nguyen SQ, Divino CM, Wang JL, Dikman SH (May 2006). "Laparoscopic management of gastrointestinal stromal tumors". Surg Endosc. 20 (5): 713–6. doi:10.1007/s00464-005-0435-8. PMID 16502196. S2CID 12838290.
- Lee, Chung-Ho; Hyun, Myung-Han; Kwon, Ye-Ji; Cho, Sung-Il; Park, Sung-Soo (2012). "Deciding Laparoscopic Approaches for Wedge Resection in Gastric Submucosal Tumors: A Suggestive Flow Chart Using Three Major Determinants". Journal of the American College of Surgeons. 215 (6): 831–840. doi:10.1016/j.jamcollsurg.2012.07.009. PMID 22951033.
- Kantarjian, HM; Wolff, RA; Koller, CA. (2011). The MD Anderson Manual of Medical Oncology (2nd ed.). McGraw-Hill. ISBN 978-0-07-170106-8.
- Wilkinson MJ, Fitzgerald JE, Strauss DC, Hayes AJ, Thomas JM, Messiou C, Fisher C, Benson C, Tekkis PP, Judson I (August 2015). "Surgical treatment of gastrointestinal stromal tumour of the rectum in the era of imatinib". Br J Surg. 102 (8): 965–71. doi:10.1002/bjs.9818. PMID 25970743. S2CID 2810885.
- Joensuu, Heikki (2012-06-01). "Adjuvant treatment of GIST: patient selection and treatment strategies". Nature Reviews. Clinical Oncology. 9 (6): 351–358. doi:10.1038/nrclinonc.2012.74. ISSN 1759-4782. PMID 22525709. S2CID 12733166.
- Joensuu, Heikki (2012-10-22). "Adjuvant therapy for high-risk gastrointestinal stromal tumour: considerations for optimal management". Drugs. 72 (15): 1953–1963. doi:10.2165/11635590-000000000-00000. ISSN 0012-6667. PMID 22994537. S2CID 43794982.
- Reichardt P, Blay JY, Boukovinas I, et al. (2012). "Adjuvant therapy in primary GIST: state-of-the-art". Annals of Oncology. 23 (11): 2776–2781. doi:10.1093/annonc/mds198. PMID 22831984.
- Cohen MH, Johnson JR, Justice R, Pazdur R (2012). "Approval summary: imatinib mesylate for one or three years in the adjuvant treatment of gastrointestinal stromal tumors. U.S. Food and Drug Administration, Silver Spring, MD 20993-0002, USA". Oncologist. 17 (7): 992–997. doi:10.1634/theoncologist.2012-0109. PMC 3399657. PMID 22643537.
- Patel Shreyaskumar R; Wong Patrick (2009). "The Efficacy of Imatinib in Unresectable/Metastatic Gastrointestinal Stromal Tumors". US Oncology. 5 (1): 61–4. doi:10.17925/ohr.2009.05.1.61. S2CID 78453531.
- Okuno, S (14 Sep 2011). "The Use of Tyrosine Kinase Inhibitors for Gastrointestinal Stromal Tumors (GIST)". Contemporary Oncology. Spring 2011. 3.
- "News: Genetic Variations in GI Tumors Determine Which Medications Are Efficacious". Genetic Engineering & Biotechnology News. 13 Nov 2008.
- ASCO-SEP 3rd ed
- "FDA approves avapritinib for gastrointestinal stromal tumor with a rare mutation". FDA. 9 January 2020.
- Avapritinib in advanced gastrointestinal stromal tumor: case series and review of the literature from a tertiary care center in India Saurav Verma, Rohit Reddy, Sheragaru Hanumanthappa Chandrashekhara, Shamim Ahmed Shamim, Sarthak Tripathy, and Sameer Rastogi Future Science OA https://www.future-science.com/doi/10.2144/fsoa-2020-0178
- Pazdur, Richard. FDA Approval for Regorafenib. National Cancer Institute.
- Pappo AS, Janeway KA (Feb 2009). "Pediatric gastrointestinal stromal tumors". Hematol Oncol Clin North Am. 23 (1): 15–34. doi:10.1016/j.hoc.2008.11.005. PMID 19248968.
- Kelly L, Bryan K, Kim SY, Janeway KA, Killian JK, Schildhaus HU, Miettinen M, Helman L, Meltzer PS, van de Rijn M, Debiec-Rychter M, O'Sullivan M (2013). "Post-Transcriptional Dysregulation by miRNAs Is Implicated in the Pathogenesis of Gastrointestinal Stromal Tumor [GIST]". PLOS ONE. 8 (5): e64102. Bibcode:2013PLoSO...864102K. doi:10.1371/journal.pone.0064102. PMC 3663836. PMID 23717541.
- Rege TA, Wagner AJ, Corless CL, Heinrich MC, Hornick JL (Apr 2011). ""Pediatric-type" gastrointestinal stromal tumors in adults: distinctive histology predicts genotype and clinical behavior". Am J Surg Pathol. 35 (4): 495–504. doi:10.1097/PAS.0b013e31820e5f7d. PMID 21358303. S2CID 40111082.
General sources
- de Silva CM, Reid R (2003). "Gastrointestinal stromal tumors (GIST): c-KIT mutations, CD117 expression, differential diagnosis and targeted cancer therapy with Imatinib" (PDF). Pathol Oncol Res. 9 (1): 13–9. doi:10.1007/BF03033708. PMID 12704441. S2CID 3814815.
- Kitamura Y, Hirota S, Nishida T (Apr 2003). "Gastrointestinal stromal tumors (GIST): a model for molecule-based diagnosis and treatment of solid tumors". Cancer Sci. 94 (4): 315–20. doi:10.1111/j.1349-7006.2003.tb01439.x. PMID 12824897. S2CID 31070671.
External links
- GIST Cancer UK
- Surgery Questions in GIST ESUN (August 15, 2006)
- SPAEN (Sarcoma Patients EuroNet) - European Network of Sarcoma, GIST and Desmoid Patient Advocacy Groups
- GIST Support International
- Life Raft Group International GIST Advocacy Organization
- American Cancer Society Patient Guide to GIST tumors.
- Cancer.Net: Gastrointestinal Stromal Tumor