Entropy of mixing
In thermodynamics, the entropy of mixing is the increase in the total entropy when several initially separate systems of different composition, each in a thermodynamic state of internal equilibrium, are mixed without chemical reaction by the thermodynamic operation of removal of impermeable partition(s) between them, followed by a time for establishment of a new thermodynamic state of internal equilibrium in the new unpartitioned closed system.
In general, the mixing may be constrained to occur under various prescribed conditions. In the customarily prescribed conditions, the materials are each initially at a common temperature and pressure, and the new system may change its volume, while being maintained at that same constant temperature, pressure, and chemical component masses. The volume available for each material to explore is increased, from that of its initially separate compartment, to the total common final volume. The final volume need not be the sum of the initially separate volumes, so that work can be done on or by the new closed system during the process of mixing, as well as heat being transferred to or from the surroundings, because of the maintenance of constant pressure and temperature.
The internal energy of the new closed system is equal to the sum of the internal energies of the initially separate systems. The reference values for the internal energies should be specified in a way that is constrained to make this so, maintaining also that the internal energies are respectively proportional to the masses of the systems.[1]
For concision in this article, the term 'ideal material' is used to refer to either an ideal gas (mixture) or an ideal solution.
In the special case of mixing ideal materials, the final common volume is in fact the sum of the initial separate compartment volumes. There is no heat transfer and no work is done. The entropy of mixing is entirely accounted for by the diffusive expansion of each material into a final volume not initially accessible to it.
In the general case of mixing non-ideal materials, however, the total final common volume may be different from the sum of the separate initial volumes, and there may occur transfer of work or heat, to or from the surroundings; also there may be a departure of the entropy of mixing from that of the corresponding ideal case. That departure is the main reason for interest in entropy of mixing. These energy and entropy variables and their temperature dependences provide valuable information about the properties of the materials.
On a molecular level, the entropy of mixing is of interest because it is a macroscopic variable that provides information about constitutive molecular properties. In ideal materials, intermolecular forces are the same between every pair of molecular kinds, so that a molecule feels no difference between other molecules of its own kind and of those of the other kind. In non-ideal materials, there may be differences of intermolecular forces or specific molecular effects between different species, even though they are chemically non-reacting. The entropy of mixing provides information about constitutive differences of intermolecular forces or specific molecular effects in the materials.
The statistical concept of randomness is used for statistical mechanical explanation of the entropy of mixing. Mixing of ideal materials is regarded as random at a molecular level, and, correspondingly, mixing of non-ideal materials may be non-random.
Mixing of ideal species at constant temperature and pressure
In ideal species, intermolecular forces are the same between every pair of molecular kinds, so that a molecule "feels" no difference between itself and its molecular neighbors. This is the reference case for examining corresponding mixing of non-ideal species.
For example, two ideal gases, at the same temperature and pressure, are initially separated by a dividing partition.
Upon removal of the dividing partition, they expand into a final common volume (the sum of the two initial volumes), and the entropy of mixing is given by


where is the gas constant, the total number of moles and the mole fraction of component , which initially occupies volume . After the removal of the partition, the moles of component may explore the combined volume , which causes an entropy increase equal to for each component gas.









In this case, the increase in entropy is entirely due to the irreversible processes of expansion of the two gases, and involves no heat or work flow between the system and its surroundings.
Gibbs free energy of mixing
The Gibbs free energy change determines whether mixing at constant (absolute) temperature and pressure is a spontaneous process. This quantity combines two physical effects—the enthalpy of mixing, which is a measure of the energy change, and the entropy of mixing considered here.



For an ideal gas mixture or an ideal solution, there is no enthalpy of mixing (), so that the Gibbs free energy of mixing is given by the entropy term only:


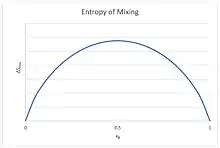
For an ideal solution, the Gibbs free energy of mixing is always negative, meaning that mixing of ideal solutions is always spontaneous. The lowest value is when the mole fraction is 0.5 for a mixture of two components, or 1/n for a mixture of n components.
Solutions and temperature dependence of miscibility
Ideal and regular solutions
The above equation for the entropy of mixing of ideal gases is valid also for certain liquid (or solid) solutions—those formed by completely random mixing so that the components move independently in the total volume. Such random mixing of solutions occurs if the interaction energies between unlike molecules are similar to the average interaction energies between like molecules.[2]: 149 [3] The value of the entropy corresponds exactly to random mixing for ideal solutions and for regular solutions, and approximately so for many real solutions.[3][4]
For binary mixtures the entropy of random mixing can be considered as a function of the mole fraction of one component.
![{\displaystyle \Delta S_{\text{mix}}=-nR(x_{1}\ln x_{1}+x_{2}\ln x_{2})=-nR[x\ln x+(1-x)\ln(1-x)]}](../I/332099383026025341c3e41b845a195998241b5a.svg)
For all possible mixtures, , so that and are both negative and the entropy of mixing is positive and favors mixing of the pure components.





Also the curvature of as a function of is given by the second derivative

This curvature is negative for all possible mixtures , so that mixing two solutions to form a solution of intermediate composition also increases the entropy of the system. Random mixing therefore always favors miscibility and opposes phase separation.

For ideal solutions, the enthalpy of mixing is zero so that the components are miscible in all proportions. For regular solutions a positive enthalpy of mixing may cause incomplete miscibility (phase separation for some compositions) at temperatures below the upper critical solution temperature (UCST).[2]: 186 This is the minimum temperature at which the term in the Gibbs energy of mixing is sufficient to produce miscibility in all proportions.

Systems with a lower critical solution temperature
Nonrandom mixing with a lower entropy of mixing can occur when the attractive interactions between unlike molecules are significantly stronger (or weaker) than the mean interactions between like molecules. For some systems this can lead to a lower critical solution temperature (LCST) or lower limiting temperature for phase separation.
For example, triethylamine and water are miscible in all proportions below 19 °C, but above this critical temperature, solutions of certain compositions separate into two phases at equilibrium with each other.[2]: 187 [5] This means that is negative for mixing of the two phases below 19 °C and positive above this temperature. Therefore, is negative for mixing of these two equilibrium phases. This is due to the formation of attractive hydrogen bonds between the two components that prevent random mixing. Triethylamine molecules cannot form hydrogen bonds with each other but only with water molecules, so in solution they remain associated to water molecules with loss of entropy. The mixing that occurs below 19 °C is due not to entropy but to the enthalpy of formation of the hydrogen bonds.


Lower critical solution temperatures also occur in many polymer-solvent mixtures.[6] For polar systems such as polyacrylic acid in 1,4-dioxane, this is often due to the formation of hydrogen bonds between polymer and solvent. For nonpolar systems such as polystyrene in cyclohexane, phase separation has been observed in sealed tubes (at high pressure) at temperatures approaching the liquid-vapor critical point of the solvent. At such temperatures the solvent expands much more rapidly than the polymer, whose segments are covalently linked. Mixing therefore requires contraction of the solvent for compatibility of the polymer, resulting in a loss of entropy.[6]
Statistical thermodynamical explanation of the entropy of mixing of ideal gases
Since thermodynamic entropy can be related to statistical mechanics or to information theory, it is possible to calculate the entropy of mixing using these two approaches. Here we consider the simple case of mixing ideal gases.
Proof from statistical mechanics
Assume that the molecules of two different substances are approximately the same size, and regard space as subdivided into a square lattice whose cells are the size of the molecules. (In fact, any lattice would do, including close packing.) This is a crystal-like conceptual model to identify the molecular centers of mass. If the two phases are liquids, there is no spatial uncertainty in each one individually. (This is, of course, an approximation. Liquids have a "free volume". This is why they are (usually) less dense than solids.) Everywhere we look in component 1, there is a molecule present, and likewise for component 2. After the two different substances are intermingled (assuming they are miscible), the liquid is still dense with molecules, but now there is uncertainty about what kind of molecule is in which location. Of course, any idea of identifying molecules in given locations is a thought experiment, not something one could do, but the calculation of the uncertainty is well-defined.
We can use Boltzmann's equation for the entropy change as applied to the mixing process

where is the Boltzmann constant. We then calculate the number of ways of arranging molecules of component 1 and molecules of component 2 on a lattice, where





is the total number of molecules, and therefore the number of lattice sites. Calculating the number of permutations of objects, correcting for the fact that of them are identical to one another, and likewise for ,


After applying Stirling's approximation for the factorial of a large integer m:
- ,

the result is
![{\displaystyle \Delta S_{mix}=-k_{\text{B}}[N_{1}\ln(N_{1}/N)+N_{2}\ln(N_{2}/N)]=-k_{\text{B}}N[x_{1}\ln x_{1}+x_{2}\ln x_{2}]}](../I/f8fd27bfea9b3f3a7d2614177ee9189dd19d9cf5.svg)
where we have introduced the mole fractions, which are also the probabilities of finding any particular component in a given lattice site.

Since the Boltzmann constant , where is the Avogadro constant, and the number of molecules , we recover the thermodynamic expression for the mixing of two ideal gases,



![{\displaystyle \Delta S_{\text{mix}}=-nR[x_{1}\ln x_{1}+x_{2}\ln x_{2}]}](../I/5015585c738e1a41032821be8d5731abdaab64db.svg)
This expression can be generalized to a mixture of components, , with




Relationship to information theory
The entropy of mixing is also proportional to the Shannon entropy or compositional uncertainty of information theory, which is defined without requiring Stirling's approximation. Claude Shannon introduced this expression for use in information theory, but similar formulas can be found as far back as the work of Ludwig Boltzmann and J. Willard Gibbs. The Shannon uncertainty is not the same as the Heisenberg uncertainty principle in quantum mechanics which is based on variance. The Shannon entropy is defined as:

where pi is the probability that an information source will produce the ith symbol from an r-symbol alphabet and is independent of previous symbols. (thus i runs from 1 to r ). H is then a measure of the expected amount of information (log pi) missing before the symbol is known or measured, or, alternatively, the expected amount of information supplied when the symbol becomes known. The set of messages of length N symbols from the source will then have an entropy of NH.
The thermodynamic entropy is only due to positional uncertainty, so we may take the "alphabet" to be any of the r different species in the gas, and, at equilibrium, the probability that a given particle is of type i is simply the mole fraction xi for that particle. Since we are dealing with ideal gases, the identity of nearby particles is irrelevant. Multiplying by the number of particles N yields the change in entropy of the entire system from the unmixed case in which all of the pi were either 1 or 0. We again obtain the entropy of mixing on multiplying by the Boltzmann constant .

So thermodynamic entropy with r chemical species with a total of N particles has a parallel to an information source that has r distinct symbols with messages that are N symbols long.
Application to gases
In gases there is a lot more spatial uncertainty because most of their volume is merely empty space. We can regard the mixing process as allowing the contents of the two originally separate contents to expand into the combined volume of the two conjoined containers. The two lattices that allow us to conceptually localize molecular centers of mass also join. The total number of empty cells is the sum of the numbers of empty cells in the two components prior to mixing. Consequently, that part of the spatial uncertainty concerning whether any molecule is present in a lattice cell is the sum of the initial values, and does not increase upon "mixing".
Almost everywhere we look, we find empty lattice cells. Nevertheless, we do find molecules in a few occupied cells. When there is real mixing, for each of those few occupied cells, there is a contingent uncertainty about which kind of molecule it is. When there is no real mixing because the two substances are identical, there is no uncertainty about which kind of molecule it is. Using conditional probabilities, it turns out that the analytical problem for the small subset of occupied cells is exactly the same as for mixed liquids, and the increase in the entropy, or spatial uncertainty, has exactly the same form as obtained previously. Obviously the subset of occupied cells is not the same at different times. But only when there is real mixing and an occupied cell is found do we ask which kind of molecule is there.
See also: Gibbs paradox, in which it would seem that "mixing" two samples of the same gas would produce entropy.
Application to solutions
If the solute is a crystalline solid, the argument is much the same. A crystal has no spatial uncertainty at all, except for crystallographic defects, and a (perfect) crystal allows us to localize the molecules using the crystal symmetry group. The fact that volumes do not add when dissolving a solid in a liquid is not important for condensed phases. If the solute is not crystalline, we can still use a spatial lattice, as good an approximation for an amorphous solid as it is for a liquid.
The Flory–Huggins solution theory provides the entropy of mixing for polymer solutions, in which the macromolecules are huge compared to the solvent molecules. In this case, the assumption is made that each monomer subunit in the polymer chain occupies a lattice site.
Note that solids in contact with each other also slowly interdiffuse, and solid mixtures of two or more components may be made at will (alloys, semiconductors, etc.). Again, the same equations for the entropy of mixing apply, but only for homogeneous, uniform phases.
Mixing under other constraints
Mixing with and without change of available volume
In the established customary usage, expressed in the lead section of this article, the entropy of mixing comes from two mechanisms, the intermingling and possible interactions of the distinct molecular species, and the change in the volume available for each molecular species, or the change in concentration of each molecular species. For ideal gases, the entropy of mixing at prescribed common temperature and pressure has nothing to do with mixing in the sense of intermingling and interactions of molecular species, but is only to do with expansion into the common volume.[7]: 273
According to Fowler and Guggenheim (1939/1965),[8] the conflating of the just-mentioned two mechanisms for the entropy of mixing is well established in customary terminology, but can be confusing unless it is borne in mind that the independent variables are the common initial and final temperature and total pressure; if the respective partial pressures or the total volume are chosen as independent variables instead of the total pressure, the description is different.
Mixing with each gas kept at constant partial volume, with changing total volume
In contrast to the established customary usage, "mixing" might be conducted reversibly at constant volume for each of two fixed masses of gases of equal volume, being mixed by gradually merging their initially separate volumes by use of two ideal semipermeable membranes, each permeable only to one of the respective gases, so that the respective volumes available to each gas remain constant during the merge. Either one of the common temperature or the common pressure is chosen to be independently controlled by the experimenter, the other being allowed to vary so as to maintain constant volume for each mass of gas. In this kind of "mixing", the final common volume is equal to each of the respective separate initial volumes, and each gas finally occupies the same volume as it did initially.[9][10]: 163–164 [11]: 217 [12][13][14]
This constant volume kind of "mixing", in the special case of perfect gases, is referred to in what is sometimes called Gibbs' theorem.[9][12][14] It states that the entropy of such "mixing" of perfect gases is zero.
Mixing at constant total volume and changing partial volumes, with mechanically controlled varying pressure, and constant temperature
An experimental demonstration may be considered. The two distinct gases, in a cylinder of constant total volume, are at first separated by two contiguous pistons made respectively of two suitably specific ideal semipermeable membranes. Ideally slowly and fictively reversibly, at constant temperature, the gases are allowed to mix in the volume between the separating membranes, forcing them apart, thereby supplying work to an external system. The energy for the work comes from the heat reservoir that keeps the temperature constant. Then, by externally forcing ideally slowly the separating membranes together, back to contiguity, work is done on the mixed gases, fictively reversibly separating them again, so that heat is returned to the heat reservoir at constant temperature. Because the mixing and separation are ideally slow and fictively reversible, the work supplied by the gases as they mix is equal to the work done in separating them again. Passing from fictive reversibility to physical reality, some amount of additional work, that remains external to the gases and the heat reservoir, must be provided from an external source for this cycle, as required by the second law of thermodynamics, because this cycle has only one heat reservoir at constant temperature, and the external provision of work cannot be completely efficient.[10]: 163–164
Gibbs' paradox: "mixing" of identical species versus mixing of closely similar but non-identical species
For entropy of mixing to exist, the putatively mixing molecular species must be chemically or physically detectably distinct. Thus arises the so-called Gibbs paradox, as follows. If molecular species are identical, there is no entropy change on mixing them, because, defined in thermodynamic terms, there is no mass transfer, and thus no thermodynamically recognized process of mixing. Yet the slightest detectable difference in constitutive properties between the two species yields a thermodynamically recognized process of transfer with mixing, and a possibly considerable entropy change, namely the entropy of mixing.
The "paradox" arises because any detectable constitutive distinction, no matter how slight, can lead to a considerably large change in amount of entropy as a result of mixing. Though a continuous change in the properties of the materials that are mixed might make the degree of constitutive difference tend continuously to zero, the entropy change would nonetheless vanish discontinuously when the difference reached zero.[15]: 87
From a general physical viewpoint, this discontinuity is paradoxical. But from a specifically thermodynamic viewpoint, it is not paradoxical, because in that discipline the degree of constitutive difference is not questioned; it is either there or not there. Gibbs himself did not see it as paradoxical. Distinguishability of two materials is a constitutive, not a thermodynamic, difference, for the laws of thermodynamics are the same for every material, while their constitutive characteristics are diverse.[16]
Though one might imagine a continuous decrease of the constitutive difference between any two chemical substances, physically it cannot be continuously decreased till it actually vanishes.[10]: 164 [17][note 1] It is hard to think of a smaller difference than that between ortho- and para-hydrogen. Yet they differ by a finite amount. The hypothesis, that the distinction might tend continuously to zero, is unphysical. This is neither examined nor explained by thermodynamics. Differences of constitution are explained by quantum mechanics, which postulates discontinuity of physical processes.[18]
For a detectable distinction, some means should be physically available. One theoretical means would be through an ideal semi-permeable membrane.[11]: 217 It should allow passage, backwards and forwards, of one species, while passage of the other is prevented entirely. The entirety of prevention should include perfect efficacy over a practically infinite time, in view of the nature of thermodynamic equilibrium. Even the slightest departure from ideality, as assessed over a finite time, would extend to utter non-ideality, as assessed over a practically infinite time. Such quantum phenomena as tunneling ensure that nature does not allow such membrane ideality as would support the theoretically demanded continuous decrease, to zero, of detectable distinction. The decrease to zero detectable distinction must be discontinuous.
For ideal gases, the entropy of mixing does not depend on the degree of difference between the distinct molecular species, but only on the fact that they are distinct; for non-ideal gases, the entropy of mixing can depend on the degree of difference of the distinct molecular species. The suggested or putative "mixing" of identical molecular species is not in thermodynamic terms a mixing at all, because thermodynamics refers to states specified by state variables, and does not permit an imaginary labelling of particles. Only if the molecular species are different is there mixing in the thermodynamic sense.[19][11]: 217–218 [20][7]: 274, 516–517 [21][22]
See also
Notes
- Partington (1949) cites Larmor (1929).
References
- Prigogine, I. (1955/1967). Introduction to Thermodynamics of Irreversible Processes, third edition, Interscience Publishers, New York, p. 12.
- Atkins, P.W., de Paula, J. (2006). Atkins' Physical Chemistry, eighth edition, W.H. Freeman, New York, ISBN 978-0-7167-8759-4.
- K. Denbigh, "The Principles of Chemical Equilibrium" (3rd ed., Cambridge University Press 1971) p.432
- P.A. Rock "Chemical Thermodynamics. Principles and Applications.(MacMillan 1969) p.263
- M.A. White, "Properties of Materials" (Oxford University Press 1999) p.175
- Cowie, J.M.G. "Polymers: Chemistry and Physics of Modern Materials" (2nd edn, Blackie 1991) p.174-176
- Bailyn, M. (1994). A Survey of Thermodynamics, American Institute of Physics, New York, ISBN 0-88318-797-3.
- Fowler, R., Guggenheim, E.A. (1939/1965). Statistical Thermodynamics. A version of Statistical Mechanics for Students of Physics and Chemistry, Cambridge University Press, Cambridge UK, pages 163-164
- Planck, M. (1897/1903). Treatise on Thermodynamics, translated with the author's sanction by Alexander Ogg, Longmans, Green and Co., London, Sections 235-236.
- Partington, J.R. (1949). An Advanced Treatise on Physical Chemistry, Volume 1, Fundamental Principles. The Properties of Gases, Longmans, Green, and Co., London.
- Adkins, C. J. (1983) [1963]. Equilibrium Thermodynamics (3rd ed.). London: McGraw-Hill. ISBN 0-521-25445-0. OCLC 9132054.
- Callen, H.B. (1960/1985). Thermodynamics and an Introduction to Thermostatistics, second edition, Wiley, New York, ISBN 981-253-185-8, pages 69-70.
- Buchdahl, H.A. (1966). The Concepts of Classical Thermodynamics, Cambridge University Press, London, pages 170-171.
- Iribarne, J.V., Godson, W.L. (1973/1981), Atmospheric Thermodynamics, second edition, D. Reidel, Kluwer Academic Publishers, Dordrecht, ISBN 90-277-1296-4, pages 48-49.
- ter Haar, D., Wergeland, H. (1966). Elements of Thermodynamics, Addison-Wesley Publishing, Reading MA.
- Truesdell, C. (1969). Rational Thermodynamics: a Course of Lectures on Selected Topics, McGraw-Hill Book Company, New York, p. 6.
- Larmor, J. (1929), Mathematical and Physical Papers, volume 2, Cambridge University Press, Cambridge UK, p. 99.
- Landé, A. (1955). Foundations of Quantum Mechanics: a Study in Continuity and Symmetry, Yale University Press, New Haven, p.10.
- Tolman, R.C. (1938). The Principles of Statistical Mechanics, Oxford University Press, Oxford, pages 626-628.
- Landsberg, P.T. (1978). Thermodynamics and Statistical Mechanics, Oxford University Press, Oxford, ISBN 0-19-851142-6, page 74.
- Grandy, W.T., Jr (2008). Entropy and the Time Evolution of Macroscopic Systems, Oxford University Press, Oxford, ISBN 978-0-19-954617-6, pages 60-62.
- Kondepudi, D. (2008). Introduction to Modern Thermodynamics, Wiley, Chichester, ISBN 978-0-470-01598-8, pages 197-199.