HH-suite
The HH-suite is an open-source software package for sensitive protein sequence searching. It contains programs that can search for similar protein sequences in protein sequence databases. Sequence searches are a standard tool in modern biology with which the function of unknown proteins can be inferred from the functions of proteins with similar sequences. HHsearch and HHblits are two main programs in the package and the entry point to its search function, the latter being a faster iteration.[2][3] HHpred is an online server for protein structure prediction that uses homology information from HH-suite.[4]
| Developer(s) | Johannes Söding, Michael Remmert, Andreas Biegert, Andreas Hauser, Markus Meier, Martin Steinegger |
|---|---|
| Stable release | 3.3.0
/ 25 August 2020 |
| Repository | |
| Written in | C++ |
| Operating system | Unix-like; Debian package available[1] |
| Available in | English |
| Type | Bioinformatics tool |
| License | GPL v3 |
| Website | https://github.com/soedinglab/hh-suite |
The HH-suite searches for sequences using hidden Markov models (HMMs). The name comes from the fact that it performs HMM-HMM alignments. Among the most popular methods for protein sequence matching, the programs have been cited more than 5000 times total according to Google Scholar.[5]
Background
Proteins are central players in all of life's processes. Understanding them is central to understanding molecular processes in cells. This is particularly important in order to understand the origin of diseases. But for a large fraction of the approximately 20 000 human proteins the structures and functions remain unknown. Many proteins have been investigated in model organisms such as many bacteria, baker's yeast, fruit flies, zebra fish or mice, for which experiments can be often done more easily than with human cells. To predict the function, structure, or other properties of a protein for which only its sequence of amino acids is known, the protein sequence is compared to the sequences of other proteins in public databases. If a protein with sufficiently similar sequence is found, the two proteins are likely to be evolutionarily related ("homologous"). In that case, they are likely to share similar structures and functions. Therefore, if a protein with a sufficiently similar sequence and with known functions and/or structure can be found by the sequence search, the unknown protein's functions, structure, and domain composition can be predicted. Such predictions greatly facilitate the determination of the function or structure by targeted validation experiments.
Sequence searches are frequently performed by biologists to infer the function of an unknown protein from its sequence. For this purpose, the protein's sequence is compared to the sequences of other proteins in public databases and its function is deduced from those of the most similar sequences. Often, no sequences with annotated functions can be found in such a search. In this case, more sensitive methods are required to identify more remotely related proteins or protein families. From these relationships, hypotheses about the protein's functions, structure, and domain composition can be inferred. HHsearch performs searches with a protein sequence through databases. The HHpred server and the HH-suite software package offer many popular, regularly updated databases, such as the Protein Data Bank, as well as the InterPro, Pfam, COG, and SCOP databases.
Algorithm
Modern sensitive methods for protein search utilize sequence profiles. They may be used to compare a sequence to a profile, or in more advanced cases such as HH-suite, to match among profiles.[2][6][7][8] Profiles and alignments are themselves derived from matches, using for example PSI-BLAST or HHblits. A position-specific scoring matrix (PSSM) profile contains for each position in the query sequence the similarity score for the 20 amino acids. The profiles are derived from multiple sequence alignments (MSAs), in which related proteins are written together (aligned), such that the frequencies of amino acids in each position can be interpreted as probabilities for amino acids in new related proteins, and be used to derive the "similarity scores". Because profiles contain much more information than a single sequence (e.g. the position-specific degree of conservation), profile-profile comparison methods are much more powerful than sequence-sequence comparison methods like BLAST or profile-sequence comparison methods like PSI-BLAST.[6]
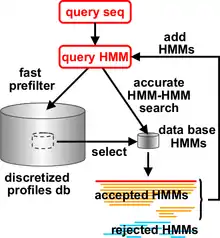
HHpred and HHsearch represent query and database proteins by profile hidden Markov models (HMMs), an extension of PSSM sequence profiles that also records position-specific amino acid insertion and deletion frequencies. HHsearch searches a database of HMMs with a query HMM. Before starting the search through the actual database of HMMs, HHsearch/HHpred builds a multiple sequence alignment of sequences related to the query sequence/MSA using the HHblits program. From this alignment, a profile HMM is calculated. The databases contain HMMs that are precalculated in the same fashion using PSI-BLAST. The output of HHpred and HHsearch is a ranked list of database matches (including E-values and probabilities for a true relationship) and the pairwise query-database sequence alignments.
HHblits, a part of the HH-suite since 2001, builds high-quality multiple sequence alignments (MSAs) starting from a single query sequence or a MSA. As in PSI-BLAST, it works iteratively, repeatedly constructing new query profiles by adding the results found in the previous round. It matches against a pre-built HMM databases derived from protein sequence databases, each representing a "cluster" of related proteins. In the case of HHblits, such matches are done on the level of HMM-HMM profiles, which grants additional sensitivity. Its prefiltering reduces the tens of millions HMMs to match against to a few thousands of them, thus speeding up the slow HMM-HMM comparison process.[3]
The HH-suite comes with a number of pre-built profile HMMs that can be searched using HHblits and HHsearch, among them a clustered version of the UniProt database, of the Protein Data Bank of proteins with known structures, of Pfam protein family alignments, of SCOP structural protein domains, and many more.[9]
Applications
Applications of HHpred and HHsearch include protein structure prediction, complex structure prediction, function prediction, domain prediction, domain boundary prediction, and evolutionary classification of proteins.[10]
HHsearch is often used for homology modeling, that is, to build a model of the structure of a query protein for which only the sequence is known: For that purpose, a database of proteins with known structures such as the protein data bank is searched for "template" proteins similar to the query protein. If such a template protein is found, the structure of the protein of interest can be predicted based on a pairwise sequence alignment of the query with the template protein sequence. For example, a search through the PDB database of proteins with solved 3D structure takes a few minutes. If a significant match with a protein of known structure (a "template") is found in the PDB database, HHpred allows the user to build a homology model using the MODELLER software, starting from the pairwise query-template alignment.
HHpred servers have been ranked among the best servers during CASP7, 8, and 9, for blind protein structure prediction experiments. In CASP9, HHpredA, B, and C were ranked 1st, 2nd, and 3rd out of 81 participating automatic structure prediction servers in template-based modeling[11] and 6th, 7th, 8th on all 147 targets, while being much faster than the best 20 servers.[12] In CASP8, HHpred was ranked 7th on all targets and 2nd on the subset of single domain proteins, while still being more than 50 times faster than the top-ranked servers.[4]
Contents
In addition to HHsearch and HHblits, the HH-suite contains programs and perl scripts for format conversion, filtering of MSAs, generation of profile HMMs, the addition of secondary structure predictions to MSAs, the extraction of alignments from program output, and the generation of customized databases.
| hhblits | (Iteratively) search an HHblits database with a query sequence or MSA |
|---|---|
| hhsearch | Search an HHsearch database of HMMs with a query MSA or HMM |
| hhmake | Build an HMM from an input MSA |
| hhfilter | Filter an MSA by maximum sequence identity, coverage, and other criteria |
| hhalign | Calculate pairwise alignments, dot plots etc. for two HMMs/MSAs |
| reformat.pl | Reformat one or many MSAs |
| addss.pl | Add Psipred predicted secondary structure to an MSA or HHM file |
| hhmakemodel.pl | Generate MSAs or coarse 3D models from HHsearch or HHblits results |
| hhblitsdb.pl | Build HHblits database with prefiltering, packed MSA/HMM, and index files |
| multithread.pl | Run a command for many files in parallel using multiple threads |
| splitfasta.pl | Split a multiple-sequence FASTA file into multiple single-sequence files |
| renumberpdb.pl | Generate PDB file with indices renumbered to match input sequence indices |
The HMM-HMM alignment algorithm of HHblits and HHsearch was significantly accelerated using vector instructions in version 3 of the HH-suite.[13]
See also
References
- Debian hhsuite package
- Söding J (2005). "Protein homology detection by HMM-HMM comparison". Bioinformatics. 21 (7): 951–960. doi:10.1093/bioinformatics/bti125. PMID 15531603.
- Remmert M, Biegert A, Hauser A, Söding J (2011). "HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment" (PDF). Nat. Methods. 9 (2): 173–175. doi:10.1038/NMETH.1818. hdl:11858/00-001M-0000-0015-8D56-A. PMID 22198341. S2CID 205420247.
- Söding J, Biegert A, Lupas AN (2005). "The HHpred interactive server for protein homology detection and structure prediction". Nucleic Acids Research. 33 (Web Server issue): W244–248. doi:10.1093/nar/gki408. PMC 1160169. PMID 15980461.
- Citations to HHpred, to HHsearch, to HHblits
- Jaroszewski L, Rychlewski L, Godzik A (2000). "Improving the quality of twilight-zone alignments". Protein Science. 9 (8): 1487–1496. doi:10.1110/ps.9.8.1487. PMC 2144727. PMID 10975570.
- Sadreyev RI, Baker D, Grishin NV (2003). "Profile–profile comparisons by COMPASS predict intricate homologies between protein families". Protein Science. 12 (10): 2262–2272. doi:10.1110/ps.03197403. PMC 2366929. PMID 14500884.
- Dunbrack RL Jr (2006). "Sequence comparison and protein structure prediction". Current Opinion in Structural Biology. 16 (3): 374–384. doi:10.1016/j.sbi.2006.05.006. PMID 16713709.
- Li, Zhaoyu. "Some Notes about HHSuite". Retrieved 3 April 2019.
- Guerler A, Govindarajoo B, Zhang Y (2013). "Mapping Monomeric Threading to Protein–Protein Structure Prediction". Journal of Chemical Information and Modeling. 53 (3): 717–25. doi:10.1021/ci300579r. PMC 4076494. PMID 23413988.
- Official CASP9 results for the template-based modeling category (121 targets)
- Official CASP9 results for all 147 targets
- Steinegger M, Meier M, Mirdita M, Vöhringer H, Haunsberger S, Söding J (2019). "HH-suite3 for fast remote homology detection and deep protein annotation". BMC Bioinformatics. 20 (1): 473. doi:10.1186/s12859-019-3019-7. PMC 6744700. PMID 31521110.
External links
- Soeding Lab at Max-Planck Institute in Göttingen - HH-suite developers
- Precompiled HH-suite binaries and databases download from developers
- HHpred — free server at Max-Planck Institute in Tuebingen
- HHblits — free server at Max-Planck Institute in Tuebingen
- CASP website
- CASP9 template-based modeling results
- HH-suite debian package
- HH-suite ubuntu package
- HH-suite arch linux user repository