Hajos–Parrish–Eder–Sauer–Wiechert reaction
The Hajos–Parrish–Eder–Sauer–Wiechert reaction in organic chemistry is a proline catalysed asymmetric aldol reaction. The reaction is named after the principal investigators of the two groups who reported it simultaneously: Zoltan Hajos and David Parrish from Hoffmann-La Roche[1][2] and Rudolf Wiechert and co-workers from Schering AG.[3] Discovered in the 1970s the original Hajos-Parrish catalytic procedure – shown in the reaction equation, leading to the optically active bicyclic ketol – paved the way of asymmetric organocatalysis. The Eder-Sauer-Wiechert modification lead directly to the optically active enedione, through the loss of water from the bicyclic ketol shown in figure.
It has been used extensively as a tool in the synthesis of steroids and other enantiomerically pure molecules.[4]
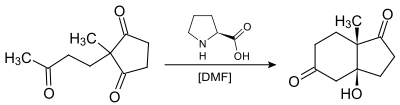
In the original reaction shown in the figure above naturally occurring chiral proline is the chiral catalyst in an Aldol reaction. The starting material is an achiral triketone and it requires just 3% of proline to obtain the reaction product, a ketol in 93% enantiomeric excess. As shown above, Hajos and Parrish worked at ambient temperature in dimethylformamide (DMF) solvent using a catalytic amount (3% molar equiv.) of (S)-(−)-proline enabling them to isolate the optically active intermediate bicyclic ketol. Thus, they described the first use of proline in a catalytic asymmetric aldol reaction.
History
Researches on asymmetric enamine catalysis applied to important intermediates in steroids synthesis is due to an increased interest for efficient and convenient steroid total syntheses in the 1960s. In particular, two industrial groups in the early 1970s reported proline-catalyzed intramolecular aldol reactions.
-7a-methyl-2%252C3%252C7%252C7a-tetrahydro-1H-indene-1%252C5(6H)-dione.svg.png.webp)
In 1971, the Schering group headed by Escher worked under non biological conditions using (S)-Proline (47 mol%), 1N perchloric acid, in acetonitrile at 80 °C. Hence, they could not isolate the Hajos-Parrish intermediate bicyclic ketol but instead the condensation product (S)-7a-methyl-2,3,7,7a-tetrahydro-1H-indene-1,5(6H)-dione through the loss of water.[5] Thirty-seven years later[6] a new group at Schering AG published the continuation of the earlier Schering work.[3] Instead of the aforementioned non biological conditions the new group used the Hajos-Parrish catalytic procedure. Thus, they could isolate the optically active 6,5-bicyclic ketol described so far only in the Hajos-Parrish publications.[1][2]
In 1974, Hajos and Parrish published the synthesis of bicyclic ketol intermediates in good yield and enantiomeric excess.[2]
They investigated further the exact configuration of the cis-fused-7a-methyl- 6,5-bicyclic-ketol shown in the reaction scheme above by circular dichroism, and these results were confirmed by a single-crystal X-ray diffraction study. The centrosymmetrical crystal of the corresponding racemic ketol without a heavy atom label has been obtained by the use of racemic proline. It showed by X-ray diffraction an axial orientation of the angular methyl group and an equatorial orientation of the hydroxyl group in the chair conformer of the six-membered ring. This is in good agreement with the crystal structure of the CD-ring of digitoxigenin.[7] The structure of this ketol and its ethyl homologue are shown as follows:
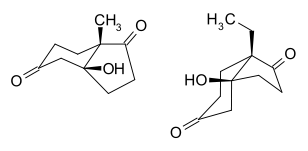
Similar studies of the 7a-ethyl-homologue showed that the ethyl bicyclic ketol existed in a cis conformation in which the 7a-ethyl group is equatorially oriented and the hydroxyl group is axially oriented in the chair form of the six-membered ring as shown above. The reason for a preference for this conformation could be enhanced 1,3-diaxial interaction in the other cis conformer between the angular ethyl group and the axial hydrogens at C-4 and C-6 in the six membered ring.
Intermolecular versions
In a 2000 study the Barbas group found that intermolecular aldol additions (those between ketones and aldehydes) are also possible albeit with use of considerably more proline:[8]
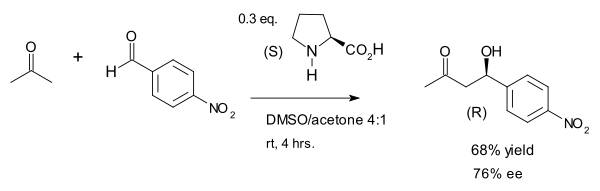
The authors noted the similarity of proline, the aldolase antibodies they had created[9] and natural aldolase enzymes aldolase A all of which operate through an enamine intermediate. In this reaction the large concentration of acetone (one of the two reactants) suppresses various possible side-reactions: reaction of the ketone with proline to an oxazolidinone and reaction of the aldehyde with proline to an azomethine ylide.
Notz and List went on to expand the utility of this reaction to the synthesis of 1,2-diols:[10]
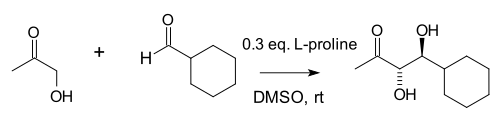
In their full account of their 2000 Communication, the group revealed that proline together with the thiazolium salt 5,5-dimethyl thiazolidinium-4-carboxylate were found to be the most effective catalysts among a large group of amines, while catalysis with (S)-1-(2-pyrrolidinylmethyl)-pyrrolidine salts formed the basis for the development of diamine organocatalysts that have proven effective in a wide variety or organocatalytic reactions.[11]
The asymmetric synthesis of the Wieland-Miescher ketone (1985) is another intramolecular reaction also based on proline, that was explored by the Barbas group in 2000.[12] In this study the Barbas group demonstrated for the first time that proline can catalyze the cascade Michael-aldol reaction through combined iminium-enamine catalysis. This work is significant because despite the 30-year history and application of the Hajos-Parrish reaction in industry, the triketone substrate for this reaction had always been synthesized in a discrete independent step, demonstrating that there was a fundamental lack of understanding of the chemical mechanism of this reaction. The Barbas group had reported the aldolase antibody catalyzed iminium-enamine Robinson annulation in their 1997 study that marked the beginning of their studies in the area now called organocatalysis.[13] In a report published in 2002 Carlos F. Barbas III said: "Work in the 1970s on proline-catalyzed intramolecular aldol addition reactions by synthetic organic chemists Zoltan G. Hajos and David R. Parrish of the chemical research department at Hoffmann-La Roche, Nutley, N.J., inspired us to look more closely at parallels between small-molecule catalysts and enzymes".[14]
In 2002 the Macmillan group was the first to demonstrate the proline catalyzed Aldol reaction between different aldehydes.[15] This reaction is unusual because in general aldehydes will self-condense.
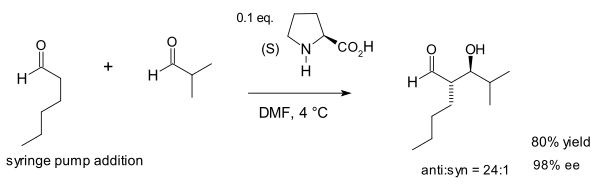
The organocatalytic intermolecular aldol reaction is now known as the Barbas-List Aldol reaction.[16]
Reaction mechanism
Several reaction mechanisms for the triketone reaction have been proposed over the years. Hajos and Parrish proposed the enamine mechanism in their paper [2]. However, their experiment with a stoichiometric amount of labeled water (H218O) supported a carbinolamine mechanism. Therefore, Hajos put forward (1974) a hemiaminal intermediate.[2] The Agami mechanism (1984) has an enamine intermediate with two proline units involved in the transition state (based on experimental reaction kinetics)[17] and according to a mechanism by Houk (2001)[18][19] a single proline unit suffices with a cyclic transition state and with the proline carboxyl group involved in hydrogen bonding.
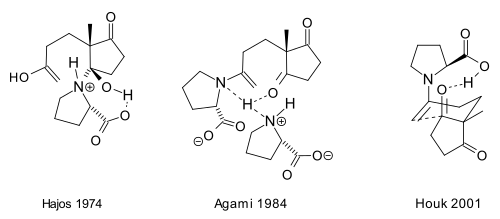
The hemiaminal (carbinolamine) put forward by Hajos in 1974 can change to a tautomeric iminium hydroxide intermediate. The iminium hydroxide ion caused enolization of the side chain methyl ketone would be followed by ring closure to the above shown optically active bicyclic ketol product (see Figure 1.) under the influence of the catalytic amount of (S)-(−)-proline. Pengxin Zhou, Long Zhang, Sanzhong Luo, and Jin-Pei Cheng obtained excellent results using the simple chiral primary amine t-Bu-CH(NH2)-CH2-NEt2.TfOH for the synthesis of both the Wieland-Miescher ketone and the Hajos-Parrish ketone as well as their analogues.[20] This supports the iminium mechanism, because it is textbook chemistry that primary amines form imines rather than enamines with carbonyl compounds.
The Hajos 1974 carbinolamine mechanism has had an unwitting support in a more recent paper by Michael Limbach.[21] The triketone starting material 2- methyl-2-(3-oxobutyl)-1,3-cyclopentanedione gave the expected optically active bicyclic ketol (+)-(3aS,7aS)-3a,4,7,7a-tetrahydro-3a-hydroxy-7a-methyl-1,5(6H)-indanedione with (S)-(−)-proline catalyst. On the other hand, the stereochemical outcome is reversed with ee selectivities of up to 83% by using the homologue amino acid catalysts, such as (S)-β-homoproline, [(pyrrolidine-(2S)-yl) acetic acid]. The virtual anomaly can be explained with a top side approach of the bulkier beta amino acids to the above triketone starting material of reflective symmetry. The top side approach results in the formation of an enantiotopic carbinolamine to give the (−)-(3aR,7aR)-3a,4,7,7a-tetrahydro-3a-hydroxy-7a-methyl-1,5(6H)-indanedione bicyclic ketol enantiomer identical to the one obtained with unnatural (R)-(+)-proline. List in 2010[22] on the other hand is perplexed and surprised that Hajos rejected the enamine mechanism, certainly in light of earlier work by Spencer in 1965 on amine catalysed aldol reactions.[23] It is interesting and surprising that Eder, Sauer and Wiechert have not attempted to explain the reaction mechanism. [3]
The reaction mechanism as proposed by the Barbas group in 2000 for the intermolecular reactions[8] is based also on enamine formation and the observed stereoselectivity based on the Zimmerman-Traxler model favoring Re-face approach. This is the same mechanism proposed by Barbas for aldolase antibodies reported by the group in 1995:
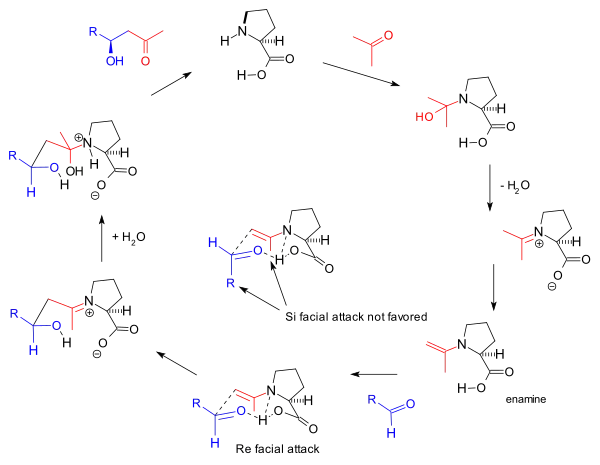
This enamine mechanism also drives the original Hajos-Parrish triketone reaction but the involvement of two proline molecules in it as proposed by Agami[17] is disputed by Barbas based on the lack of a non-linear effects[11] and supported by later studies of List based on reaction kinetics.[24] The general mechanism is further supported by List by the finding that in a reaction carried out in labeled water (H218O), the oxygen isotope finds its way into the reaction product.[25] The Hajos and Parrish experiment with a stoechiometric amount of labeled water (H218O) supported the carbinolamine mechanism.[2]
In the same study [20] the reaction of proline with acetone to the oxazolidinone (in DMSO) was examined:
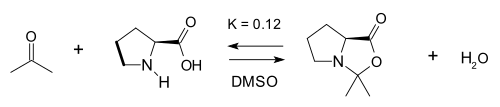
The equilibrium constant for this reaction is only 0.12 leading List to conclude that the involvement of oxazolidinone is only parasitic.
Blackmond in 2004 also found oxazolidinones as intermediates (NMR) in a related proline-catalysed α-aminooxylation of propanal with nitrosobenzene:[26]
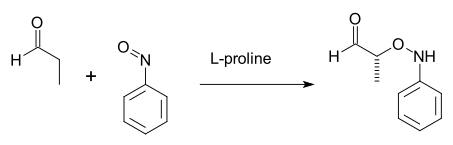
Chiong Teck Wong of the Institute of High Performance Computing Singapore studied the similar oxyamination reaction of nitrosobenzene with butanal using a chiral prolinol silyl ether catalyst.[27] His studies strongly suggest that the catalyst generates the enol, and forms an enol-catalyst complex. Nitsosobenzene subsequently reacts with the enol-catalyst complex to afford the (S)-N-nitroso aldol product in agreement with Pauling’s chart of electronegativity. Sodiumborohydride reduction of the primarily formed aldol products gave the corresponding alcohols in good yield and excellent enantioselectivity in the ratio of PN/PO=>99:1 as shown in the Scheme below. Wong suggests that the reaction mechanism of the (S)-Cat catalyzed N-nitroso aldol reaction between nitrosobenzene and butanal proceeds via an enol intermediate and not via an enamine intermediate.
2009-811.svg.png.webp)
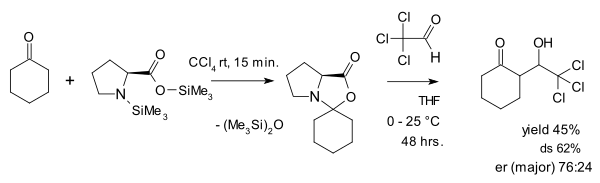
The view of oxazolidinones as a parasitic species is contested by Seebach and Eschenmoser who in 2007 published an article[28] in which they argue that oxazolidinones in fact play a pivotal role in proline catalysis. One of the things they did was reacting an oxazolidinone with the activated aldehyde chloral in an aldol addition:
In 2008, Barbas in an essay addressed the question why it took until the year 2000 before interest regenerated for this seemingly simple reaction 30 years after the pioneering work by Hajos and Parrish and why the proline catalysis mechanism appeared to be an enigma for so long.[29] One explanation has to do with different scientific cultures: a proline mechanism in the context of aldolase catalysis already postulated in 1964 by a biochemist[30] was ignored by organic chemists. Another part of the explanation was the presumed complexity of aldolase catalysis that dominated chemical thinking for a long time. Finally, research did not expand in this area at Hoffmann-La Roche after the resignation of ZGH in November, 1970.
Origin of the name of the reaction
The name for this reaction took some time to develop. In 1985 Professor Agami and associates were the first to name the proline catalyzed Robinson annulation the Hajos-Parrish reaction.[31] In 1986 Professor Henri B. Kagan and Professor Agami[32] still called it the Hajos-Parrish reaction in the Abstract of this paper. In 2001 Kagan published a paper entitled "Nonlinear Effects in Asymmetric Catalysis: A Personal Account" in Synlett.[33] In this paper he introduced the new title the Hajos-Parrish-Wiechert reaction. In 2002 Benjamin List added two more names and introduced the term Hajos–Parrish–Eder–Sauer–Wiechert reaction.[34] Scientific papers published as late as 2008 in the field of organocatalysis use either the 1985, 2001 or 2002 names of the reaction. A June, 2014 search limited to the years 2009–2014 by Google Scholar returns 44 hits for Hajos-Parrish reaction, 3 for Hajos-Parrish-Wiechert reaction and 184 for Hajos–Parrish–Eder–Sauer–Wiechert reaction. The term 'Hajos-Parrish ketone' (and similar) remains common, however.
References
- Z. G. Hajos, D. R. Parrish, German Patent DE 2102623 1971
- Hajos, Zoltan G.; Parrish, D. R. (1974). "Asymmetric synthesis of bicyclic intermediates of natural product chemistry". The Journal of Organic Chemistry. 39 (12): 1615–1621. doi:10.1021/jo00925a003.
- Eder, Ulrich (1971). "New Type of Asymmetric Cyclization to Optically Active Steroid CD Partial Structures". Angewandte Chemie International Edition in English. 10 (7): 496–497. doi:10.1002/anie.197104961.
- Wang, Zerong (15 September 2010). Comprehensive Organic Name Reactions and Reagents. Hoboken, NJ, USA: John Wiley & Sons, Inc. p. 1306. doi:10.1002/9780470638859. ISBN 978-0-470-63885-9.
- List, Benjamin (2002). "Proline-catalyzed asymmetric reactions". Tetrahedron. 58 (28): 5573–5590. doi:10.1016/S0040-4020(02)00516-1.
- Kennedy, Jason W. J.; Vietrich, Sophia; Weinmann, Hilmar; Brittain, Dominic E. A. (2008). "Synthesis of 7a-Substituted Hajos−Wiechert Ketone Analogues". The Journal of Organic Chemistry. 73 (13): 5151–5154. doi:10.1021/jo800638s. PMID 18540678.
- The crystal structure of digitoxigenin, Karle, I.L., and Karle, J., Acta Crystallogr. B, 25: 434–442 (1969).
- List, Benjamin; Lerner, Richard A.; Barbas, Carlos F. (26 February 2000). "Proline-Catalyzed Direct Asymmetric Aldol Reactions". Journal of the American Chemical Society. American Chemical Society (ACS). 122 (10): 2395–2396. doi:10.1021/ja994280y. ISSN 0002-7863.
- Wagner, J; Lerner, RA; Barbas, CF (December 1995). "Efficient aldolase catalytic antibodies that use the enamine mechanism of natural enzymes". Science. 270 (5243): 1797–800. Bibcode:1995Sci...270.1797W. doi:10.1126/science.270.5243.1797. PMID 8525368. S2CID 12714361.
- Notz, Wolfgang; List, Benjamin (14 July 2000). "Catalytic Asymmetric Synthesis of anti-1,2-Diols". Journal of the American Chemical Society. American Chemical Society (ACS). 122 (30): 7386–7387. doi:10.1021/ja001460v. ISSN 0002-7863.
- Sakthivel, Kandasamy (2001). "Amino Acid Catalyzed Direct Asymmetric Aldol Reactions: A Bioorganic Approach to Catalytic Asymmetric Carbon−Carbon Bond-Forming Reactions". Journal of the American Chemical Society. 123 (22): 5260–5267. doi:10.1021/ja010037z. PMID 11457388.
- Bui, Tommy (2000). "A proline-catalyzed asymmetric Robinson annulation reaction". Tetrahedron Letters. 41 (36): 6951–6954. doi:10.1016/S0040-4039(00)01180-1.
- Zhong, Guofu (1997). "Antibody-Catalyzed Enantioselective Robinson Annulation". Journal of the American Chemical Society. 119 (34): 8131–8132. doi:10.1021/ja970944x.
- Science & Technology, February 2002, Volume 80, Number 8, CENEAR 80 08 p. 33 ISSN 0009-2347
- Northrup, Alan B. (2002). "The First Direct and Enantioselective Cross-Aldol Reaction of Aldehydes". Journal of the American Chemical Society. 124 (24): 6798–6799. doi:10.1021/ja0262378. PMID 12059180.
- Ramachary, Dhevalapally B. (2009). "Direct Catalytic Asymmetric Synthesis of Highly Functionalized 2-Methylchroman-2,4-diols via Barbas-List Aldol Reaction". Chemistry - A European Journal. 15 (18): 4516–4522. doi:10.1002/chem.200900066. PMID 19308984.
- Agami, Claude (1984). "Stereochemistry-59". Tetrahedron. 40 (6): 1031–1038. doi:10.1016/S0040-4020(01)91242-6.
- Bahmanyar, S. (2001). "The Origin of Stereoselectivity in Proline-Catalyzed Intramolecular Aldol Reactions". Journal of the American Chemical Society. 123 (51): 12911–12912. doi:10.1021/ja011714s. PMID 11749554.
- Bahmanyar, S. (2001). "Transition States of Amine-Catalyzed Aldol Reactions Involving Enamine Intermediates: Theoretical Studies of Mechanism, Reactivity, and Stereoselectivity". Journal of the American Chemical Society. 123 (45): 11273–11283. doi:10.1021/ja011403h. PMID 11697970.
- Zhou, Pengxin; Zhang, Long; Luo, Sanzhong; Cheng, Jin-Pei (14 February 2012). "Asymmetric Synthesis of Wieland–Miescher and Hajos–Parrish Ketones Catalyzed by an Amino-Acid-Derived Chiral Primary Amine". The Journal of Organic Chemistry. American Chemical Society (ACS). 77 (5): 2526–2530. doi:10.1021/jo202433v. ISSN 0022-3263. PMID 22316216.
- β-Homoamino acids as catalysts on enantioselective intra- and intermoelcular aldol reactions by Michael Limbach, Tetrahedron Letters 47 (2006) 3843–3847
- List, B. (2010). "Emil Knoevenagel and the Roots of Aminocatalysis". Angewandte Chemie International Edition in English. 49 (10): 1730–1734. doi:10.1002/anie.200906900. PMID 20175175.
- Spencer, T. (1965). "Observations on amine catalysis of formation and dehydration of ketols". Tetrahedron Letters. 6 (43): 3889–3897. doi:10.1016/S0040-4039(01)89143-7. PMID 5842468.
- Hoang, Linh (2003). "Kinetic and Stereochemical Evidence for the Involvement of Only One Proline Molecule in the Transition States of Proline-Catalyzed Intra- and Intermolecular Aldol Reactions". Journal of the American Chemical Society. 125 (1): 16–17. doi:10.1021/ja028634o. PMID 12515489.
- List, B. (2004). "Asymmetric Catalysis Special Feature Part II: New mechanistic studies on the proline-catalyzed aldol reaction". Proceedings of the National Academy of Sciences. 101 (16): 5839–5842. Bibcode:2004PNAS..101.5839L. doi:10.1073/pnas.0307979101. PMC 395996. PMID 15073330.
- Iwamura, Hiroshi (2004). "Probing the Active Catalyst in Product-Accelerated Proline-Mediated Reactions". Journal of the American Chemical Society. 126 (50): 16312–16313. doi:10.1021/ja0444177. PMID 15600319.
- A theoretical investigation on the mechanism of the alpha,alpha-diphenylprolinol trimethylsilyl ether-catalyzed oxyamination reaction, Chiong Teck Wong, Tetrahedron Letters 50 (2009) 811–813.
- Are Oxazolidinones Really Unproductive, Parasitic Species in Proline Catalysis? – Thoughts and Experiments Pointing to an Alternative View Helvetica Chimica Acta Volume 90, Issue 3, Date: March 2007, Pages: 425–471 Dieter Seebach, Albert K. Beck, D. Michael Badine, Michael Limbach, Albert Eschenmoser, Adi M. Treasurywala, Reinhard Hobi, Walter Prikoszovich, Bernard Linder doi:10.1002/hlca.200790050
- Organocatalysis Lost: Modern Chemistry, Ancient Chemistry, and an Unseen Biosynthetic Apparatus Carlos F. Barbas III Angew. Chem. Int. Ed. 2008, 47, 42–47 doi:10.1002/anie.200702210
- Rutter, W. J. (1964). "Evolution of Aldolase". Fed. Proc. 23: 1248–57. PMID 14236133.
- Agami, Claude (1985). "A new diagnostic tool for elucidating the mechanism of enantioselective reactions. Application to the Hajos–Parrish reaction". J. Chem. Soc., Chem. Commun. (8): 441–442. doi:10.1039/c39850000441.
- Gilman, Henry; Jones, R. G. (1940). "Triphenylindium1". Journal of the American Chemical Society. 62 (9): 2353–2357. doi:10.1021/ja01866a025.
- Synlett 2001, No. SI, 888–899
- List, Benjamin (2002). "Proline-catalyzed asymmetric reactions". Tetrahedron. 58 (28): 5573–5590. doi:10.1016/s0040-4020(02)00516-1.