Haploinsufficiency
Haploinsufficiency in genetics describes a model of dominant gene action in diploid organisms, in which a single copy of the wild-type allele at a locus in heterozygous combination with a variant allele is insufficient to produce the wild-type phenotype. Haploinsufficiency may arise from a de novo or inherited loss-of-function mutation in the variant allele, such that it yields little or no gene product (often a protein). Although the other, standard allele still produces the standard amount of product, the total product is insufficient to produce the standard phenotype. This heterozygous genotype may result in a non- or sub-standard, deleterious, and (or) disease phenotype. Haploinsufficiency is the standard explanation for dominant deleterious alleles.
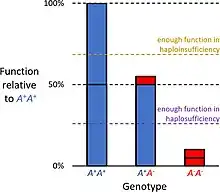
In the alternative case of haplosufficiency, the loss-of-function allele behaves as above, but the single standard allele in the heterozygous genotype produces sufficient gene product to produce the same, standard phenotype as seen in the homozygote. Haplosufficiency accounts for the typical dominance of the "standard" allele over variant alleles, where the phenotypic identity of genotypes heterozygous and homozygous for the allele defines it as dominant, versus a variant phenotype produced only by the genotype homozygous for the alternative allele, which defines it as recessive.
Mechanism
The alteration in the gene dosage, which is caused by the loss of a functional allele, is also called allelic insufficiency.
Haploinsufficiency in humans
About 3,000 human genes cannot tolerate loss of one of the two alleles.[1]
An example of this is seen in the case of Williams syndrome, a neurodevelopmental disorder caused by the haploinsufficiency of genes at 7q11.23. The haploinsufficiency is caused by the copy-number variation (CNV) of 28 genes led by the deletion of ~1.6 Mb. These dosage-sensitive genes are vital for human language and constructive cognition.[2]
Another example is the haploinsufficiency of telomerase reverse transcriptase which leads to anticipation in autosomal dominant dyskeratosis congenita. It is a rare inherited disorder characterized by abnormal skin manifestations, which results in bone marrow failure, pulmonary fibrosis and an increased predisposition to cancer. A null mutation in motif D of the reverse transcriptase domain of the telomerase protein, hTERT, leads to this phenotype. Thus telomerase dosage is important for maintaining tissue proliferation.[3]
A variation of haploinsufficiency exists for mutations in the gene PRPF31, a known cause of autosomal dominant retinitis pigmentosa. There are two wild-type alleles of this gene—a high-expressivity allele and a low-expressivity allele. When the mutant gene is inherited with a high-expressivity allele, there is no disease phenotype. However, if a mutant allele and a low-expressivity allele are inherited, the residual protein levels falls below that required for normal function, and disease phenotype is present.[4]
Copy number variation (CNV) refers to the differences in the number of copies of a particular region of the genome. This leads to too many or too few of the dosage sensitive genes. The genomic rearrangements, that is, deletions or duplications, are caused by the mechanism of non-allelic homologous recombination (NAHR). In the case of the Williams Syndrome, the microdeletion includes the ELN gene. The hemizygosity of the elastin is responsible for supravalvular aortic stenosis, the obstruction in the left ventricular outflow of blood in the heart.[5][6]
Other examples include:
- Some cancers
- 1q21.1 deletion syndrome
- 5q- syndrome in myelodysplastic syndrome (MDS)
- 22q11.2 deletion syndrome
- CHARGE syndrome
- Cleidocranial dysostosis
- Ehlers–Danlos syndrome
- Frontotemporal dementia caused by mutations in progranulin
- GLUT1 deficiency (DeVivo syndrome)[7]
- Haploinsufficiency of A20
- Holoprosencephaly caused by haploinsufficiency in the Sonic Hedgehog gene
- Holt–Oram syndrome
- Marfan syndrome[8]
- Phelan–McDermid syndrome
- Polydactyly
- Dravet Syndrome
- ZTTK
- FOXP1 Syndrome
- NR4A2-related syndrome
Methods of detection
The most direct method to detect haploinsufficiency is the heterozygous deletion of one allele in a model organism. This can be done in tissue culture cells or in single-celled organisms such as yeast (Saccharomyces cerevisiae).[9]
References
- Bartha, István; di Iulio, Julia; Venter, J. Craig; Telenti, Amalio (January 2018). "Human gene essentiality". Nature Reviews Genetics. 19 (1): 51–62. doi:10.1038/nrg.2017.75. ISSN 1471-0056. PMID 29082913. S2CID 9025172.
- Tassabehji, M.; Metcalfe, K.; Karmiloff-Smith, A.; Carette, M. J.; Grant, J.; Dennis, N.; Reardon, W.; Splitt, M.; Read, A. P.; Donnai, D. (January 1999). "Williams syndrome: use of chromosomal microdeletions as a tool to dissect cognitive and physical phenotypes". American Journal of Human Genetics. 64 (1): 118–125. doi:10.1086/302214. ISSN 0002-9297. PMC 1377709. PMID 9915950.
- Armanios, M.; et al. (2004). "Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenital". Genetics. 102 (44): 15960–15964.
- McGee, TL; Devoto, M; Ott, J; et al. (November 1997). "Evidence that the penetrance of mutations at the RP11 locus causing dominant retinitis pigmentosa is influenced by a gene linked to the homologous RP11 allele". Am J Hum Genet. 61 (5): 1059–66. doi:10.1086/301614. PMC 1716046. PMID 9345108.
- Lee, J. A.; Lupski, J. R. (2006). "Genomic rearrangements and gene copy-number alterations as a cause of nervous system disorders". Neuron. 52 (52): 103–121. doi:10.1016/j.neuron.2006.09.027. PMID 17015230. S2CID 22412305.
- Menga, X.; Lub, X.; Morrisc, C.A.; Keating, M.T. (1998). "A Novel Human GeneFKBP6Is Deleted in Williams Syndrome*1". Genomics. 52 (52): 130–137. doi:10.1006/geno.1998.5412. PMID 9782077.
- Rotstein M, Engelstad K, Yang H; et al. (2010). "Glut1 deficiency: inheritance pattern determined by haploinsufficiency". Ann Neurol. 68 (6): 955–8. doi:10.1002/ana.22088. PMC 2994988. PMID 20687207.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - Robinson, P. N.; Arteaga-Solis, E.; Baldock, C.; Collod-Béroud, G.; Booms, P.; De Paepe, A.; Dietz, H. C.; Guo, G.; Handford, P. A.; Judge, D. P.; Kielty, C. M.; Loeys, B.; Milewicz, D. M.; Ney, A.; Ramirez, F. (2006-03-29). "The molecular genetics of Marfan syndrome and related disorders". Journal of Medical Genetics. 43 (10): 769–787. doi:10.1136/jmg.2005.039669. ISSN 1468-6244. PMC 2563177. PMID 16571647.
- Strome, Erin D.; Wu, Xiaowei; Kimmel, Marek; Plon, Sharon E. (March 2008). "Heterozygous screen in Saccharomyces cerevisiae identifies dosage-sensitive genes that affect chromosome stability". Genetics. 178 (3): 1193–1207. doi:10.1534/genetics.107.084103. ISSN 0016-6731. PMC 2278055. PMID 18245329.
Further reading
- Ebert, BL; et al. (2008). "Identification of RPS14 as a 5q- syndrome gene by RNA interference screen". Nature. 451 (451): 335–340. Bibcode:2008Natur.451..335E. doi:10.1038/nature06494. PMC 3771855. PMID 18202658.
- Griffiths, Anthony J.; et al. (200). Introduction to Genetic Analysis (8th ed.). W.H. Freeman. ISBN 0-7167-4939-4.