USH1C
Harmonin is a protein that in humans is encoded by the USH1C gene.[5][6][7] It is expressed in sensory cells of the inner ear and retina, where it plays a role in hearing, balance, and vision.[5][6][8][9][10] Mutations at the USH1C locus cause Usher syndrome type 1c and nonsyndromic sensorineural deafness.[5][6][8][11]




Gene and protein structure
The USH1C gene is located on chromosome 11 and contains 28 exons.[5] Alternative splicing generates multiple mRNA transcript variants, some of which are associated with the rare disorder phenotypes of Usher syndrome and nonsyndromic sensorineural deafness.[5][6] The encoded protein harmonin has multiple protein isoforms due to the alternative splicing, including a standard isoform with 552 amino acids.[5] Harmonin contains a PDZ domain, which assists in attaching the protein to the cell membrane and to cytoskeletal components.[5]
Inner ear function
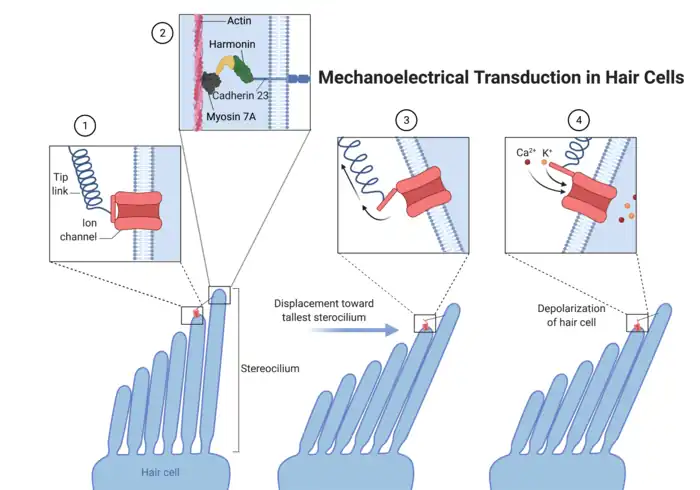
Harmonin is found at the apex of inner hair cells (IHCs), which convert mechanical signals from sound waves into electrical signals interpreted by the brain as sound.[5][9][10][12] IHCs have an apical bundle of actin-rich stereocilia that vary in height and are connected to each other by flexible tip links.[5][9][10][12] Tip links are protein complexes of cadherin 23 (CDH23) and protocadherin 15 (PCDH15).[5][9][10][12] Harmonin binds to proteins that are involved in connecting the tip link to the cytoskeleton.[13][14][15] Sound waves physically displace the bundle towards the tallest stereocilium, stretching the tip links and causing mechanically gated ion channels to open.[15] Influx of calcium (Ca2+) and potassium (K+) depolarizes the hair cell, triggering the release of excitatory neurotransmitters onto the innervating nerve terminals.[15] The process is called mechanoelectrical transduction and ultimately results in the perception of sound.[15] Intact tip links and their associated proteins, including harmonin, are required for channel activation and normal hearing.[5][12]
Mutations
USH1C mutations inherited in an autosomal recessive pattern have been identified as the genetic basis of both Usher syndrome type 1c and nonsyndromic sensorineural deafness type 18 (DFNB18).[5][6][8][11] A diploid individual has two alleles, or copies, of the USH1C gene, one inherited from the maternal parent and one inherited from the paternal parent.[11] A wild type USH1C allele encodes the functional harmonin protein, whereas a mutant USH1C allele cannot.[11] Expression of the wild type USH1C allele is dominant over the mutant USH1C allele.[11] An individual with two wild type alleles will be unaffected, an individual with one wild type allele and one mutant allele will be an asymptomatic carrier, and an individual with two mutant alleles will experience the disorder phenotype.[11] The molecular personality of each USH1c mutation determines whether the resulting phenotype is nonsyndromic deafness or Usher syndrome.[6][11]
A common mutation that causes Usher syndrome is a single nucleotide polymorphism (SNP) at nucleotide 216 that replaces the base guanine with the base adenine, creating a frameshift with a deletion of 35 base pairs.[16][17] The 216 G to A mutation introduces a cryptic splice site that is used instead of the wild-type splice site during post-transcriptional RNA processing.[16][17] The consequent mis-splicing causes the 35-nucleotide deletion in the mature mRNA transcript.[16][17] Since the change in the RNA sequence is not a multiple of three, the mRNA contains a frameshift and a premature stop codon after 189 nucleotides.[16][17] If the mRNA were translated, a 135-amino-acid protein would be formed instead of wild type harmonin, but there is no evidence that protein is made from the misspliced mRNA.[16][17] An individual will experience Usher syndrome type 1c if they are homozygous for the 216 G to A mutant allele, which is found at high frequencies in Acadian populations.[16]
Usher syndrome
Usher syndrome is a rare autosomal recessive disorder caused by a mutation in one of several genes involved in hearing, balance, and vision.[11] There are multiple types of Usher syndrome that vary in severity and symptomatology depending on the affected gene.[11] Usher syndrome type 1c is caused by a mutation at the USH1C locus and is characterized by childhood onset of bilateral sensorineural hearing loss, vestibular dysfunction, and vision loss from retinitis pigmentosa.[5][6][8][11] Usher syndrome type 1 is the most severe form of Usher syndrome.[18] The prevalence of Usher syndrome is approximately 3-6 in 100,000 live births, rendering the disorder the most common cause of comorbid hearing and vision loss.[11][18] Usher syndrome type 1c is prevalent in Acadian populations but is found worldwide.[6][16][19] Although there is no cure, studies to evaluate potential gene therapies are ongoing.[17][19]
Gene therapy
Human hearing develops by 19 weeks gestation.[20] At birth, individuals with Usher syndrome type 1c already have sensorineural hearing loss from mutant harmonin, and mammalian hearing loss is presently irreversible.[17] It is hypothesized that gene therapy to correct the USH1C mutation and restore the wild type harmonin protein is most effective during the critical developmental window that is hypothesized to close one week before hearing onset.[17][21] Studies of mouse models of Usher syndrome type 1c note that hearing develops in mice at postnatal day 12.[22] Gene therapy to deliver an antisense oligonucleotide to the mouse inner ear rescued wild type harmonin mRNA splicing as well as hearing and vestibular function when delivered at embryonic day 12.5 or postnatal days 1-5 but was significantly less effective thereafter.[17] The antisense oligonucleotide sequence is complementary to a segment of the 216 G to A mutant mRNA and mechanically blocks the cryptic splice site so that the wild type splice site is used.[17] Likewise, gene therapy to deliver an adeno-associated viral (AAV) vector encoding wild type harmonin to the mouse inner ear rescued hearing and vestibular function when delivered on postnatal days 0-1 but was ineffective at postnatal days 10-12.[23]
Gene therapy is controversial due to ethical and social considerations.[24][25][26] For example, some members of the deaf community embrace hearing loss as a positive aspect of their identity and culture that they do not wish to change, whereas other members seek therapeutic interventions.[24][25] However, there is widespread interest in developing gene therapies to provide treatment options for patients, especially when the symptoms of a genetic disorder are debilitating and difficult to manage with conventional strategies.[19][26]
References
- GRCh38: Ensembl release 89: ENSG00000006611 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000030838 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, Salem N, et al. (September 2000). "A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C". Nature Genetics. 26 (1): 51–55. doi:10.1038/79171. PMID 10973247. S2CID 9383331.
- Ahmed ZM, Smith TN, Riazuddin S, Makishima T, Ghosh M, Bokhari S, et al. (June 2002). "Nonsyndromic recessive deafness DFNB18 and Usher syndrome type IC are allelic mutations of USHIC". Human Genetics. 110 (6): 527–531. doi:10.1007/s00439-002-0732-4. PMID 12107438. S2CID 24276167.
- "Entrez Gene: USH1C Usher syndrome 1C (autosomal recessive, severe)".
- Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, et al. (September 2000). "A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene". Nature Genetics. 26 (1): 56–60. doi:10.1038/79178. PMID 10973248. S2CID 2237489.
- Gregory FD, Bryan KE, Pangršič T, Calin-Jageman IE, Moser T, Lee A (August 2011). "Harmonin inhibits presynaptic Cav1.3 Ca²⁺ channels in mouse inner hair cells". Nature Neuroscience. 14 (9): 1109–1111. doi:10.1038/nn.2895. PMC 3164920. PMID 21822269.
- Gregory FD, Pangrsic T, Calin-Jageman IE, Moser T, Lee A (July 2013). "Harmonin enhances voltage-dependent facilitation of Cav1.3 channels and synchronous exocytosis in mouse inner hair cells". The Journal of Physiology. 591 (13): 3253–3269. doi:10.1113/jphysiol.2013.254367. PMC 3717226. PMID 23613530.
- Millán JM, Aller E, Jaijo T, Blanco-Kelly F, Gimenez-Pardo A, Ayuso C (2010-12-23). "An update on the genetics of usher syndrome". Journal of Ophthalmology. 2011: 417217. doi:10.1155/2011/417217. PMC 3017948. PMID 21234346.
- Grillet N, Xiong W, Reynolds A, Kazmierczak P, Sato T, Lillo C, et al. (May 2009). "Harmonin mutations cause mechanotransduction defects in cochlear hair cells". Neuron. 62 (3): 375–387. doi:10.1016/j.neuron.2009.04.006. PMC 2691393. PMID 19447093.
- Boëda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, et al. (December 2002). "Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle". The EMBO Journal. 21 (24): 6689–6699. doi:10.1093/emboj/cdf689. PMC 139109. PMID 12485990.
- Siemens J, Kazmierczak P, Reynolds A, Sticker M, Littlewood-Evans A, Müller U (November 2002). "The Usher syndrome proteins cadherin 23 and harmonin form a complex by means of PDZ-domain interactions". Proceedings of the National Academy of Sciences of the United States of America. 99 (23): 14946–14951. Bibcode:2002PNAS...9914946S. doi:10.1073/pnas.232579599. PMC 137525. PMID 12407180.
- Yu IM, Planelles-Herrero VJ, Sourigues Y, Moussaoui D, Sirkia H, Kikuti C, et al. (June 2017). "Myosin 7 and its adaptors link cadherins to actin". Nature Communications. 8 (1): 15864. Bibcode:2017NatCo...815864Y. doi:10.1038/ncomms15864. PMC 5493754. PMID 28660889.
- Lentz J, Savas S, Ng SS, Athas G, Deininger P, Keats B (February 2005). "The USH1C 216G-->A splice-site mutation results in a 35-base-pair deletion". Human Genetics. 116 (3): 225–227. doi:10.1007/s00439-004-1217-4. PMID 15578223. S2CID 31987210.
- Wang L, Kempton JB, Jiang H, Jodelka FM, Brigande AM, Dumont RA, et al. (May 2020). "Fetal antisense oligonucleotide therapy for congenital deafness and vestibular dysfunction". Nucleic Acids Research. 48 (9): 5065–5080. doi:10.1093/nar/gkaa194. PMC 7229850. PMID 32249312.
- Reiners J, Nagel-Wolfrum K, Jürgens K, Märker T, Wolfrum U (July 2006). "Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease". Experimental Eye Research. 83 (1): 97–119. doi:10.1016/j.exer.2005.11.010. PMID 16545802.
- de Joya EM, Colbert BM, Tang PC, Lam BL, Yang J, Blanton SH, et al. (April 2021). "Usher Syndrome in the Inner Ear: Etiologies and Advances in Gene Therapy". International Journal of Molecular Sciences. 22 (8): 3910. doi:10.3390/ijms22083910. PMC 8068832. PMID 33920085.
- Locher H, Frijns JH, van Iperen L, de Groot JC, Huisman MA, Chuva de Sousa Lopes SM (October 2013). "Neurosensory development and cell fate determination in the human cochlea". Neural Development. 8 (1): 20. doi:10.1186/1749-8104-8-20. PMC 3854452. PMID 24131517.
- Ponnath A, Depreux FF, Jodelka FM, Rigo F, Farris HE, Hastings ML, Lentz JJ (February 2018). "Rescue of Outer Hair Cells with Antisense Oligonucleotides in Usher Mice Is Dependent on Age of Treatment". Journal of the Association for Research in Otolaryngology. 19 (1): 1–16. doi:10.1007/s10162-017-0640-x. PMC 5783922. PMID 29027038.
- Shnerson A, Willott JF (February 1980). "Ontogeny of the acoustic startle response in C57BL/6J mouse pups". Journal of Comparative and Physiological Psychology. 94 (1): 36–40. doi:10.1037/h0077648. PMID 7372853.
- Pan B, Askew C, Galvin A, Heman-Ackah S, Asai Y, Indzhykulian AA, et al. (March 2017). "Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c". Nature Biotechnology. 35 (3): 264–272. doi:10.1038/nbt.3801. PMC 5340578. PMID 28165476.
- Katz S (2020). "Why deaf people oppose using gene editing to "cure" deafness". Discover Magazine. Retrieved 2022-05-08.
- Mullin E (2021-01-26). "Gene therapy could end deafness. Should it?". Future Human. Retrieved 2022-05-08.
- Gonçalves GA, Paiva RM (2017). "Gene therapy: advances, challenges and perspectives". Einstein. 15 (3): 369–375. doi:10.1590/S1679-45082017RB4024. PMC 5823056. PMID 29091160.
Further reading
- Maruyama K, Sugano S (January 1994). "Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides". Gene. 138 (1–2): 171–174. doi:10.1016/0378-1119(94)90802-8. PMID 8125298.
- Suzuki Y, Yoshitomo-Nakagawa K, Maruyama K, Suyama A, Sugano S (October 1997). "Construction and characterization of a full length-enriched and a 5'-end-enriched cDNA library". Gene. 200 (1–2): 149–156. doi:10.1016/S0378-1119(97)00411-3. PMID 9373149.
- Scanlan MJ, Chen YT, Williamson B, Gure AO, Stockert E, Gordan JD, et al. (May 1998). "Characterization of human colon cancer antigens recognized by autologous antibodies". International Journal of Cancer. 76 (5): 652–658. doi:10.1002/(SICI)1097-0215(19980529)76:5<652::AID-IJC7>3.0.CO;2-P. PMID 9610721.
- Jain PK, Lalwani AK, Li XC, Singleton TL, Smith TN, Chen A, et al. (June 1998). "A gene for recessive nonsyndromic sensorineural deafness (DFNB18) maps to the chromosomal region 11p14-p15.1 containing the Usher syndrome type 1C gene". Genomics. 50 (2): 290–292. doi:10.1006/geno.1998.5320. PMID 9653658.
- Saouda M, Mansour A, Bou Moglabey Y, El Zir E, Mustapha M, Chaib H, et al. (August 1998). "The Usher syndrome in the Lebanese population and further refinement of the USH2A candidate region". Human Genetics. 103 (2): 193–198. doi:10.1007/s004390050806. PMID 9760205. S2CID 6040572.
- Scanlan MJ, Williamson B, Jungbluth A, Stockert E, Arden KC, Viars CS, et al. (April 1999). "Isoforms of the human PDZ-73 protein exhibit differential tissue expression". Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression. 1445 (1): 39–52. doi:10.1016/s0167-4781(99)00033-0. PMID 10209257.
- Kobayashi I, Imamura K, Kubota M, Ishikawa S, Yamada M, Tonoki H, et al. (October 1999). "Identification of an autoimmune enteropathy-related 75-kilodalton antigen". Gastroenterology. 117 (4): 823–830. doi:10.1016/S0016-5085(99)70340-9. PMID 10500064.
- Scanlan MJ, Gordan JD, Williamson B, Stockert E, Bander NH, Jongeneel V, et al. (November 1999). "Antigens recognized by autologous antibody in patients with renal-cell carcinoma". International Journal of Cancer. 83 (4): 456–464. doi:10.1002/(SICI)1097-0215(19991112)83:4<456::AID-IJC4>3.0.CO;2-5. PMID 10508479.
- Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, et al. (September 2000). "A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene". Nature Genetics. 26 (1): 56–60. doi:10.1038/79178. PMID 10973248. S2CID 2237489.
- Zwaenepoel I, Verpy E, Blanchard S, Meins M, Apfelstedt-Sylla E, Gal A, Petit C (2001). "Identification of three novel mutations in the USH1C gene and detection of thirty-one polymorphisms used for haplotype analysis". Human Mutation. 17 (1): 34–41. doi:10.1002/1098-1004(2001)17:1<34::AID-HUMU4>3.0.CO;2-O. PMID 11139240. S2CID 10069513.
- Ishikawa S, Kobayashi I, Hamada J, Tada M, Hirai A, Furuuchi K, et al. (April 2001). "Interaction of MCC2, a novel homologue of MCC tumor suppressor, with PDZ-domain Protein AIE-75". Gene. 267 (1): 101–110. doi:10.1016/S0378-1119(01)00378-X. PMID 11311560.
- Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, et al. (July 2001). "Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F". American Journal of Human Genetics. 69 (1): 25–34. doi:10.1086/321277. PMC 1226045. PMID 11398101.
- Savas S, Frischhertz B, Pelias MZ, Batzer MA, Deininger PL, Keats BB (January 2002). "The USH1C 216G-->A mutation and the 9-repeat VNTR(t,t) allele are in complete linkage disequilibrium in the Acadian population". Human Genetics. 110 (1): 95–97. doi:10.1007/s00439-001-0653-7. PMID 11810303. S2CID 39688683.
- Ouyang XM, Xia XJ, Verpy E, Du LL, Pandya A, Petit C, et al. (July 2002). "Mutations in the alternatively spliced exons of USH1C cause non-syndromic recessive deafness". Human Genetics. 111 (1): 26–30. doi:10.1007/s00439-002-0736-0. PMID 12136232. S2CID 8137208.
- Siemens J, Kazmierczak P, Reynolds A, Sticker M, Littlewood-Evans A, Müller U (November 2002). "The Usher syndrome proteins cadherin 23 and harmonin form a complex by means of PDZ-domain interactions". Proceedings of the National Academy of Sciences of the United States of America. 99 (23): 14946–14951. Bibcode:2002PNAS...9914946S. doi:10.1073/pnas.232579599. PMC 137525. PMID 12407180.
- Boëda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, et al. (December 2002). "Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle". The EMBO Journal. 21 (24): 6689–6699. doi:10.1093/emboj/cdf689. PMC 139109. PMID 12485990.
- Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Lainé S, et al. (March 2003). "Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin". Human Molecular Genetics. 12 (5): 463–471. doi:10.1093/hmg/ddg051. PMID 12588794.
External links
- GeneReviews/NCBI/NIH/UW entry on Usher Syndrome Type I
- USH1C human gene location in the UCSC Genome Browser.
- USH1C human gene details in the UCSC Genome Browser.
- Overview of all the structural information available in the PDB for UniProt: Q9Y6N9 (Human Harmonin) at the PDBe-KB.
- Overview of all the structural information available in the PDB for UniProt: Q9ES64 (Mouse Harmonin) at the PDBe-KB.
PDB gallery | |
|---|---|
|